Archives: NNT Reviews

Adjuvant Antibiotic Therapy After Incision and Drainage of Cutaneous Abscesses

Benefits in NNT
14
1 in 14 were helped (treatment failure prevented)
10
1 in 10 were helped (recurrence prevented)

Harms in NNT
23
1 in 23 were harmed (experienced adverse events)
View As:
Source
Gottlieb M, Demott JM, Hallock M, Peksa GD. Systemic Antibiotics for the Treatment of Skin and Soft Tissue Abscesses: A Systematic Review and Meta-Analysis. Annals of Emergency Medicine 2019;73(1):8–16.Study Population: Four studies with 2406 adults and children undergoing incision and drainage for cutaneous abscess.
Efficacy Endpoints
Treatment failure, recurrenceHarm Endpoints
Adverse events, diarrheaNarrative
Annually more than 3 million patients present to U.S. emergency departments (EDs) with cutaneous abscess, a number that has been increasing.1 Standard treatment involves incision and drainage (I&D), while routine use of systemic antibiotics after incision and drainage is controversial. Recently, two large studies found increased cure rates with systemic antibiotics after I&D compared to placebo.2, 3 The goal of the systematic review summarized here is to provide updated evidence on the efficacy of systemic antibiotics with activity against methicillin-resistant Staphylococcus aureus after I&D of cutaneous abscess.4The review identified four randomized trials comprised of 2406 adult and pediatric subjects who presented with acute, simple, cutaneous abscesses that required I&D. Three took place exclusively in the ED and one in a mix of ED and outpatient settings. In three trials, participants were randomized to receive trimethoprim–sulfamethoxazole (TMP-SMX) or placebo while one trial randomized participants to receive TMP-SMX, clindamycin, or placebo. The primary outcome was treatment failure within 21 days based on clinical assessment and the need for further intervention. Secondary outcomes were recurrence, overall adverse events (gastrointestinal symptoms, rashes, and generalized symptoms), and diarrhea.
Antibiotic therapy was associated with an increased rate of clinical cure (absolute risk difference [ARD]: 7.4%; odds ratio [OR] 2.3; 95% confidence interval [CI], 1.8 - 3.1; NNT 14) and a reduced risk of recurrence (ARD: 10%; OR: 0.3; CI: 0.2 - 0.4; NNT: 10). Antibiotic therapy was also associated with an increase in adverse events (ARD: 4.4%; OR: 1.3; CI: 1.1 - 1.6; NNH: 23) but no significant change in diarrhea.
Caveats
Caveats: The quality of evidence was high, risk of bias was low, and there was no significant heterogeneity. Additionally, another systematic review and meta-analysis, which included RCTs of antibiotics without activity against MRSA, reached the same conclusions as the authors of this analysis.5 There are however limitations. One limitation is that I&D technique was not standardized in two of the studies.6, 7 This is unlikely to have affected the outcome since I&D is a simple procedure and the two studies that did standardize the I&D technique both nonetheless demonstrated a benefit to antibiotics.2, 3 Another is that two different antibiotics (TMP-SMX and clindamycin) and multiple dosing regimens were used, though the clinical cure rate between antibiotics was not different.3 Additionally, the studies were not powered to detect rare adverse events such as severe allergic reactions and Clostridium difficile infection. Finally, there was variation in follow-up period with three studies assessing patient outcomes at 7-10 days, and the fourth study assessing outcomes at 14-21 days.4Notably, the clinical cure rate without antibiotics was 84% compared to 92% with antibiotics, and treatment failure rarely results in life-threatening complications or even hospitalization—usually just a return visit with an additional I&D and outpatient antibiotics. The slightly increased clinical cure rate must be balanced against the harms associated with antibiotic use including adverse events and antibiotic resistance.8, 9 The harms that would be caused to the community by increasing antibiotic resistance may outweigh the benefits to the individual in many cases.
In summary, adjuvant antibiotics given routinely after I&D of cutaneous abscesses were associated with increased clinical cure, decreased recurrence, and increased adverse events in this review. The benefits should be weighed against the adverse events, the cost of treatment failure, and the impact on society of increasing antibiotic usage. Based on the continued necessity for clinicians to weigh the benefits and harms of adjuvant antibiotics, the most appropriate rating is Yellow (benefits and harms should be individualized).
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
John Conway, BS; Benjamin Friedman, MDSupervising Editor: Shahriar Zehtabchi, MD
Published/Updated
October 16, 2019References:
Prochlorperazine for Treatment of Acute Migraines in Adults
Benefits in NNT
3
1 in 3 were helped (pain relief, compared to placebo)
Harms in NNT
8
1 in 8 were harmed (adverse events, compared to placebo)
View As:
Source
Golikhatir I, Cheraghmakani H, Bozorgi F, et al. The efficacy and safety of prochlorperazine in patients with acute migraine: a systemic review and meta-analysis. Headache. 2019;0:1- 19.Study Population: 5 studies of 223 adult adults with acute migraine headache
Efficacy Endpoints
Resolution of headache or reduced severityHarm Endpoints
Adverse events (akathisia, dystonia, drowsiness, and orthostatic hypotension)Narrative
Migraine headache results in over 1 million emergency department visits per year in the United States.1, 2, 3 Several treatments have been offered to treat the headache.4, 5, 6 Prochlorperazine has been tested in randomized trials and, despite adverse effects such as dystonic reactions several societies recommend its use.4, 6The systematic review and meta-analysis summarized here evaluated trials of adult patients with acute migraine who were randomized to receive prochlorperazine, placebo, or a comparator agent.7 The systematic review’s primary outcome included the number of patients with complete headache relief or reduced severity within 2 hours. This was defined by absence of headache, 30% reduction in severity, reduced severity by 2.5 out of 10 scale, or no request for rescue analgesia. As a secondary outcome, the systematic review assessed the rates of adverse events (i.e. akathisia, dystonia, drowsiness, and orthostatic hypotension).
The systematic review identified 11 moderate-to-high quality trials (771 patients), but only 5 studies (223 patients) compared prochlorperazine to placebo. Out of these five trials, two used a descriptive scale, 2 used a visual analog scale, and 1 trial used a verbal rating scale for grading severity of pain. The mean age for the enrolled patient was approximated 30 years old, and the majority of patients were female. When compared to placebo, prochlorperazine was more effective for controlling the headache (Odds ratio [OR]: 7.2, 95% confidence interval [CI]: 3.8- 13.7; Absolute risk difference [ARD]: 43%; Number needed to treat [NNT]: 3, low statistical heterogeneity, moderate to high quality of evidence). The analysis reported similar effectiveness for pain control at 60 minutes and 120 minutes after drug administration. However, prochlorperazine was associated with increased risk of adverse events compared to placebo (OR: 5.79, 95% CI 2.4-13.8; ARD: 11.4%; Number needed to harm [NNH]: 8).
Caveats
Based on this meta-analysis, prochlorperazine provided better migraine relief in adult patients than placebo. However, there are several limitations. Most studies evaluated the intravenous route, which is common in the emergency department. Studies evaluating other routes were small, and the systematic review was unable to draw clear conclusions regarding nonintravenous routes.The systematic review also analyzed the data from trials that compared prochlorperazine to other agents (ketorolac, metoclopramide, hydromorphone, ergotamine, octreotide, sumatriptan). Unfortunately, most trials were small. In general, however, prochlorperazine appeared to be more effective than other agents.
Despite co-treatment with diphenhydramine in many studies, the risk of extrapyramidal adverse events was significantly higher in patients allocated to prochlorperazine when compared with placebo (and of other comparators). However, several studies did not consistently report adverse events, and some reported adverse events in one study arm but not the other. Due to these factors, it is unclear if the reported data truly reflect the risk of adverse event associated with the use of prochlorperazine. The readers should interpret these results with caution. As expected from relatively small overall sample size, many point estimates had wide confidence intervals.
In summary, prochlorperazine seems more effective than placebo for acute migraine relief. However, it is likely associated with high risk of adverse events, including extrapyramidal symptoms. Because of the small sample size of the included trials and inconsistency in reporting the adverse events, we have assigned a color recommendation of Yellow (unclear if benefits outweigh harms) to this treatment. While prochlorperazine can effectively treat acute migraines in adults, the risk of adverse effects must be considered, as there are several other safer options for managing the headache in these patients.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Brit Long, MD; Alex Koyfman, MD; Michael Gottlieb, MD, RDMSSupervising Editor: Shahriar Zehtabchi, MD
Published/Updated
References:
Prevalence of Pulmonary Embolism in Patients Presenting With Syncope
Benefits in NNT
125
1 in 125 emergency department patients with syncope were found to have a PE
100
1 in 100 hospitalized patients with syncope were found to have a PE
Harms in NNT
Not applicable: data on harms were not available
View As:
Source
Oqab Z, Ganshorn H, Sheldon R. Prevalence of pulmonary embolism in patients presenting with syncope. A systematic review and meta-analysis. Am J Emerg Med 2018;36:551–5.Study Population: 6,608 ED patients (nine studies) and 975 hospitalized patients (three studies) presenting with syncope
Efficacy Endpoints
Prevalence of PE in patients presenting to the ED with syncopeHarm Endpoints
No harm endpoints were assessedNarrative
Syncope accounts for 1% to 3% of emergency department (ED) visits and 1% to 6% of hospital admissions.1, 2 There are numerous etiologies, ranging from relatively benign vasovagal syncope to dangerous dysrhythmias. The ED evaluation and management of syncope is composed of history, examination, and typically an electrocardiogram, with further investigation dependent on clinical decision making and suspected conditions.2 Previously, pulmonary embolism (PE) was thought to account for a small minority of patients with syncope. However, a recent study by Prandoni and colleagues3 reported a high prevalence of PE in admitted patients with syncope (3.8% of ED patients and 17.3% of hospitalized patients). Evaluating for PE in all patients with syncope carries significant risks including radiation exposure, contrast-induced nephropathy, and adverse events from anticoagulation therapy.4, 5 In this evidence-based review, we summarize and critically appraise a published meta-analysis that evaluated the overall prevalence of PE in patients presenting with syncope to provide guidance to clinicians regarding testing decisions in this population.6This meta-analysis included studies evaluating patients with syncope who presented to the ED or were admitted to the hospital that reported underlying etiologies, which included PE. There were no limitations on age, language, time, or setting, and to assess methodologic quality, authors modified an existing quality scale.6 The authors identified 1,920 studies, of which 12 papers (excluding Prandoni et al.) met inclusion criteria. Nine studies (n = 6,608 patients) took place in the ED, and three studies (n = 975 patients) occurred in the hospital environment. Weighted median age in ED patients was 61.5 years, compared to 67.1 years in hospitalized patients. PE was confirmed through computerized tomography angiography (CTA) of chest, ventilation perfusion scan, pulmonary angiography, or autopsy.
Results of the current meta-analysis suggest a low prevalence of PE in patients presenting with syncope: 0.8% (95% confidence interval [CI] = 0.5%–1.3%) in ED patients (number needed to screen = 125) and 1.0% (95% CI = 0.5%–1.9%) in hospitalized patients (number needed to screen = 100), with an overall prevalence of 0.9% (95% CI = 0.6%–1.3%).6
Caveats
The meta-analysis discussed here had several important limitations. First, the authors included both prospective and retrospective data. Additionally, only four of the included studies discussed specific diagnostic strategies for PE in this meta-analysis.6 Another concern is that the authors utilized their own modified scale to assess methodologic quality, rather than using one of the more established tools, such as QUADAS-2 or the Newcastle-Ottawa criteria.7 Moreover, the decision to order CTA was mostly based on clinician judgment. Finally, the presenting symptoms, patient characteristics, and rationale for obtaining the CTA were not discussed in most of the included trials. While CTA of chest with contrast possesses high sensitivity and specificity for diagnosis of PE in low pretest probability patients, test characteristics decrease in patients with high pretest probability.8 Discordance among radiologists for diagnosis of PE can also be severe, with poor interreader reliability.9A second important consideration is that syncope has a significant number of potential etiologies, and determining a specific cause can be difficult. Therefore, as expected, clinical heterogeneity among the included studies was significant.6 Since the studies did not systematically screen for PE, it is unclear how many cases may have been missed. Follow-up for patients discharged from the ED to ensure they did not have PE was unclear in the majority of studies. Studies also demonstrated variable patient populations and baseline characteristics.6 Most importantly, whether identifying these positive cases of PE affected long-term outcomes (e.g., mortality) of the patients is not known. PE can be asymptomatic and/or an incidental finding. A significant portion of patients demonstrate incidental PE at the time of autopsy, with rates ranging from 9% to 63%.5 Thus, PE may occur and resolve without clinical effect.
Another major caveat for diagnostic evaluation of syncope patients for PE is establishing causality. To cause syncope, a pulmonary blood clot must result in dysrhythmia, acute right ventricular failure, or a Bezold-Jarisch reflex.10 The literature suggests only PE located in the main pulmonary or lobar arteries are associated with syncope.10 However, in the study by Prandoni et al.,3 approximately one-third of PE were segmental or subsegmental, which would be unlikely to result in syncope. Therefore, it is unclear whether the diagnosed PEs were associated with the syncope or incidental findings. Additionally, it is unclear how many cases were false positives due to imaging artifact.
Based on the low prevalence of PE in patients with syncope in this meta-analysis (low-quality evidence), dedicated testing for PE in all syncope patients is not recommended. Overtesting for PE may result in risks from the testing itself, as well as side effects from anticoagulation given in cases with false-positive test results or clinically insignificant cases. We assign testing for PE in all syncope patients red (harm > benefit). While consideration of PE in patients with syncope is warranted, the decision to trigger diagnostic evaluation for PE should be guided by proper risk stratification using history and physical examination.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Brit Long, MD; Alex Koyfman, MD; Michael Gottlieb, MD, RDMSSupervising Editor: Shahriar Zehtabchi, MD
Published/Updated
October 1, 2019References:
Risk of Recurrent Venous Thromboembolism and Bleeding in Cancer Patients Treated with Direct Oral Anticoagulants Versus Low Molecular Weight Heparin
Benefits in NNT
41
1 in 41 were helped (recurrent VTE prevented)
Harms in NNT
No one was harmed (no significant difference in risk of major bleeding compared to LMWH)
View As:
Source
Dong Y, Wang Y, Ma RL, Liu M, Gao JZ, Su WY, et al. Efficacy and safety of direct oral anticoagulants versus low-molecular-weight heparin in patients with cancer: a systematic review and meta-analysis. J Thromb Thrombolysis. 2019 May 6. doi: 10.1007/s11239-019- 01871-4.Study Population: 11 studies of 4509 patients (1868 patients receiving DOACs and 2641 patients receiving LMWH)
Efficacy Endpoints
Recurrent VTEHarm Endpoints
Major bleedingNarrative
Venous thromboembolism (VTE) occurs in up to 30% of patients with cancer.1, 2 Prior guidelines have recommended low molecular weight heparin (LMWH) for 3-6 months as first-line therapy in cancer patients with newly-diagnosed VTE.3, 4, 5 Unfortunately, LMWH is associated with poor compliance due to the need for subcutaneous injection.6, 7 Direct oral anticoagulants (DOACs) have been increasingly used for the treatment of VTE, are administered orally with no requirement for regular laboratory monitoring, and may have fewer drug-drug interactions as compared with warfarin, despite DOACs possessing a greater cost compared to other therapies. Several more recent guidelines, including the National Comprehensive Cancer Network (NCCN) and International Society on Thrombosis and Haemostasis (ISTH), recommend DOACs, based on limited data.8, 9 However, other guidelines including the American Society of Clinical Oncology still preferentially recommend LMWH,10 and DOAC efficacy and safety remain controversial in patients with cancer and acute VTE when compared to LMWH.This systematic review and meta-analysis included studies comparing DOACs with LMWH for the treatment of VTE in patients with cancer.11 The primary outcomes were VTE recurrence and major bleeding in patients with cancer receiving DOACs or LMWH. Major bleeding was defined as clinically overt bleeding associated with a decrease in hemoglobin of 2 g/dL or more, requiring transfusion of two or more units of blood, occurring in a critical site (e.g. intracranial, intraspinal, intraocular, retroperitoneal, intra-articular, pericardial or intramuscular with compartment syndrome), or fatal bleeding as per ISTH criteria. Authors conducted subgroup analyses based on study design, specific medication, and duration of follow-up.
The authors identified 11 relevant studies (n=4509), with 2 randomized controlled trials (RCTs) (1 trial each evaluating edoxaban and rivaroxaban) and 9 observational cohort studies (6 studies evaluating rivaroxaban and 3 studies other DOACs). The follow-up period was ≥ 1 month in all studies. DOACs reduced VTE recurrence from 11.45% to 9.01% (absolute risk reduction [ARR] 2.44%), with a relative risk (RR) of 0.72 (95% confidence interval [CI] 0.60-0.85) and number needed to treat (NNT) of 41 compared to LMWH. Subgroup analyses of RCTs and observational studies demonstrated a consistent reduction in VTE recurrence with DOACs. Overall, there was no statistically significant difference in major bleeding, including intracerebral, retroperitoneal, and intraspinal. Subgroup analyses of only observational studies, length of follow-up (6 and 12 months), and rivaroxaban also revealed no increased risk of bleeding. However, subgroup analysis of only RCTs did find increased risk of major bleeding with DOACs (RR 1.78, 95% CI 1.11-2.87).
Caveats
This meta-analysis possesses several limitations. The 2 RCTs demonstrating increased rates of major bleeding with DOACs primarily involved the gastrointestinal (GI) tract.12, 13 However, both studies included a large number of patients with GI malignancies, and these studies were industry sponsored. It is unclear whether DOACs may be safer in patients without GI tract malignancies, and further data are needed. The definition of active cancer was not consistent in the included studies, and not all studies classified the cancer types or stages. Many studies also did not specify the type or the chronicity of VTE. Included studies evaluated different DOACs and LMWH comparators. This meta-analysis included predominately observational studies, which can introduce confounders and selection bias. While these studies demonstrated incidences of recurrent VTE similar to RCTs, differences in the baseline characteristics of patients and potential unidentified confounders can introduce bias. Follow-up and duration of therapy varied in the included studies, producing a potential source of heterogeneity.This meta-analysis suggests cancer patients who receive DOACs have significantly reduced risk of VTE when compared to LMWH, with the best evidence found with rivaroxaban. The risk of major bleeding is less clear, as data across all studies fail to show a difference, but RCT data suggest increased harm. In the context of this study, DOACs remain a viable option to reduce risk of VTE in cancer patients, particularly among patients at low risk of bleeding. Their oral administration and lack of required monitoring is patient-centric and likely improves compliance.14 We have assigned a color recommendation of Yellow (Unclear if Benefits) based upon the benefit for reduction of VTE, but potential increased risk of major bleeding reported in RCTs. Larger, high-quality RCTs are needed to establish with more certainty the promising benefits suggested by these data, as well as further study of the effects of cancer type and specific DOAC medication.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Brit Long, MD; Alex Koyfman, MD; Michael Gottlieb, MD, RDMSSupervising Editor: James McCormack, MD; Gary Green, MD
Published/Updated
References:
Rocuronium vs. Succinylcholine for Rapid Sequence Intubation
Benefits in NNT
N/A (No difference when succinylcholine is compared to recommended dose of rocuronium)
Harms in NNT
Not reported
View As:
Source
Tran DTT, Newton EK, Mount VAH, et al. Rocuronium vs. succinylcholine for rapid sequence intubation: a Cochrane systematic review. Anaesthesia. 2017;72:765-777.Study Population: 4151 adults and children of any age who underwent rapid sequence intubation, electively or emergently, across 50 studies.
Efficacy Endpoints
‘Excellent’ and ‘clinically acceptable’ intubating conditions using the Goldberg scaleHarm Endpoints
Not reportedNarrative
Rapid sequence intubation (RSI), placing a tube into the trachea facilitated by rapid sedation and paralysis to improve ventilation and oxygenation, is a common procedure in emergent, critical care, and operating room settings. There is great interest in drugs that improve the process. The two most commonly used paralytic agents in the emergency department are succinylcholine (depolarizing) and rocuronium (non-depolarizing). Traditionally succinylcholine has been the preferred muscle relaxant for RSI because of its rapid onset of 40 to 60 seconds and a short duration of action lasting 6 to 10 minutes. However, succinylcholine’s depolarizing action may lead to hyperkalemia, possibly inducing fatal cardiac arrhythmia. As a result, it is contraindicated in patients with known hyperkalemia, severe burns (beyond 48 hours), major crush injuries (beyond 48 hours), denervation syndromes, and muscular dystrophy.1 Rocuronium however, is a steroid-based non-depolarizing muscle relaxant, which has been proposed for creating intubating conditions similar to those of succinylcholine. The duration of action is longer, lasting 37-72 minutes and has an antidote, while the only contraindication is allergy.1The Cochrane review summarized here1 determines whether rocuronium creates intubating conditions comparable to those of succinylcholine, by comparing the Goldberg scale (Table 1). This scale allocates a score (1 through 4) for each of the following item: ease of intubation, vocal cord movement, and patient response to intubation. This scale gives a total point value of 12, in which three represents excellent, four to six represents good, seven to nine represents poor, and 10 to 12 represents inadequate intubating condition.
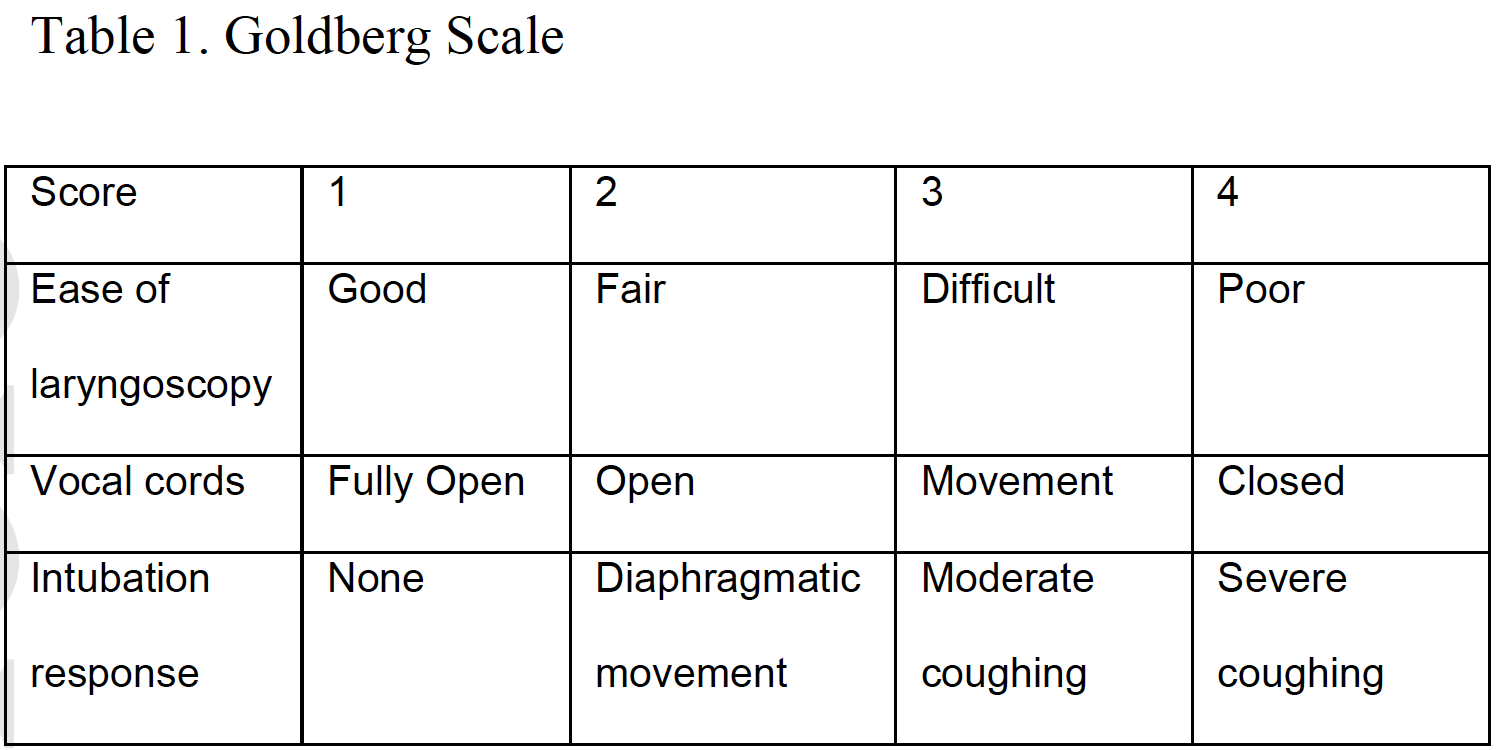
The Cochrane review1 included randomized controlled trials (RCT) and controlled clinical trials (CCT) meeting the following inclusion criteria: 1) Score of intubation is reported as the main outcome. 2) Compared succinylcholine with rocuronium. 3) The dose of rocuronium was at least 0.6 mg/kg (0.6-1.2 mg/kg) and the dose of succinylcholine was at least 1 mg/kg. The sedative agents used for induction were thiopental, benzodiazipines, propofol, etomidate, or ketamine. It is important to note that the majority of included trials were conducted in non-emergent settings and rocuronium was used at low doses (0.6-0.7 mg/kg) in most trials.
Overall, the meta-analysis revealed that succinylcholine was superior to rocuronium for achieving excellent intubating conditions (Relative Risk [RR]: 0.86, 95% Confidence Interval [CI], 0.81 - 0.92; Absolute Risk Reduction [ARR] 12%; Number-Needed-To-Treat [NNT]: 8), and for clinically acceptable conditions (RR: 0.97, 95% CI, 0.95 - 0.99; ARR: 5%; NNT 19). Heterogeneity among trials for both endpoints was very high. However, when dosing of the medication was analyzed, succinylcholine was superior only to low dose (0.6 - 0.7 mg/kg) rocuronium and there was no difference in outcome between the groups when the recommended higher dose (0.9 - 1.0 mg/kg) of rocuronium was used. Since the recommended dose for rocuronium in RSA is higher than the dose used in the Cochrane’s main analysis,2,3 we did not include the efficacy endpoints for the low-dose rocuronium in the summary table.
Caveats
The safety of rapid sequence intubation is sought by providers who have long dealt with periprocedural complications and general instability with this high-stakes procedure. It is important to note, that the Cochrane review included only 5 studies (1,073 participants) occurring in the emergency setting. Therefore, the findings of the systematic review might not be applicable to emergency department. Additionally, measuring the endpoint of “excellent” and “clinically acceptable” intubating conditions has an uncertain clinical relevance to emergency physicians due to its subjectivity and potential for bias. A more important outcome is first-pass success along with peri-intubation adverse events, such as hypoxia, hypotension, esophageal intubation, etc.The vast majority of studies in the Cochrane review compared succinylcholine with low-dose rocuronium (0.6-0.7 mg/kg). When using rocuronium, quality intubating conditions are achieved with higher doses (>0.9 mg/kg). Whereas, lower doses may take a longer onset of action resulting in the possibility of delayed/failed endotracheal tube placement or compromising the quality.
Since the publication of this Cochrane review in 2015, another study by April et al.4 based on registry data, has been published in 2018. This study included 4,275 intubations from the National Emergency Airway Registry (NEAR); comparing first-pass success rates and adverse events between succinylcholine and rocuronium. This analysis showed no difference in first-pass success (87.0% versus 87.5%) or adverse events (14.7% versus 14.8%) between succinylcholine and rocuronium groups. Moreover, the mean dose of succinylcholine was 1.8 mg/kg, whereas the mean dose of rocuronium was 1.2 mg/kg. These findings confirm the results in the subgroup analysis of the Cochrane review that compared succinylcholine with high-dose rocuronium.
Preferring one agent based on time of onset and duration of action is common and debated. Rocuronium is longer acting and has a reversal agent. Many ED physicians have more experience using succinylcholine, which is shorter acting, making a reversal agent less often helpful. Some clinicians opt for rocuronium to avoid adverse reactions (e.g. hyperkalemia), and to have the option of reversal on demand. Others recommend succinylcholine, preferring shorter paralysis.
Notably, heterogeneity in the primary and secondary outcome analyses was very high. This suggests these results should be interpreted with caution.
In summary, we believe that comparing succinylcholine with suboptimal, low doses of rocuronium is inappropriate. We have based our finding of no difference, and our color assignment (Yellow—further research needed) on the results of the proper comparison. We look forward to clinical trials comparing high dose rocuronium (>0.9 mg/kg) with succinylcholine for RSI in the emergency department setting, while focusing on relevant outcomes of first-pass success rates and adverse outcomes.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Abdullah Bakhsh, MBBS FAAEMSupervising Editor: Shahriar Zehtabchi, MD
Published/Updated
References:
Non-Invasive Positive Pressure Ventilation for Exacerbation of Chronic Obstructive Pulmonary Disease
Benefits in NNT
12
1 in 12 had death prevented
5
1 in 5 had endotracheal intubation prevented
Harms in NNT
9
1 in 9 had discomfort leading to discontinuation of treatment
3
1 in 3 had minor complications such as ear pain or skin damage
View As:
Source
Osadnik CR, Tee VS, Carson-chahhoud KV, Picot J, Wedzicha JA, Smith BJ. Non-invasive ventilation for the management of acute hypercapnic respiratory failure due to exacerbation of chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2017;7:CD004104.Study Population: Adults with hypercapneic respiratory failure due to exacerbation of chronic obstructive pulmonary disease.
Efficacy Endpoints
Death, endotracheal intubation, hospital length of stay, discomfort leading to discontinuation of treatment, minor complicationsHarm Endpoints
Discomfort leading to inability to tolerate the mask, minor complicationsNarrative
Chronic obstructive pulmonary disease (COPD) is a progressive lung disease characterized by hyperinflation of the lungs, including emphysema and chronic bronchitis. COPD death rates in the United States have declined since 1999 though predominantly in males.1 Endotracheal intubation with ventilatory support for severe exacerbations is very difficult to reverse. Noninvasive positive pressure ventilation (NIPPV), if effective in avoiding endotracheal intubation, could therefore save lives and reduce suffering.Of the seventeen studies in the Cochrane review1 all were randomized trials with parallel group design, comparing usual care plus NIPPV to usual care alone, with some variations in usual care. NIPPV was delivered via face mask, nasal mask, or either based on preference.1 A total of 1264 adults with severe exacerbation and hypercapnic respiratory failure (pH <7.35, PCO2 >45mmHg) were enrolled.
NIPPV reduced both primary outcomes including death (odds ratio 0.4, 95%CI 0.3-0.5; absolute difference 8.4%, NNT 12), and endotracheal intubation (OR 0.4, 95%CI 0.3-0.5; difference 22%, NNT 5). Length of hospital stay was also shorter with NIPPV (mean difference 3 days, 95% CI 1-6).1
Inability to comply with treatment was higher in the NIPPV group (absolute difference 11%, 95%CI 4-17%, NNH 9). Complications due to NIPPV, which included ear pain, skin breaks due to the mask, and other minor issues, occurred in nearly 1 in 3 (NNH 3). However overall complications unrelated to NIPPV were less in the two studies reporting this outcome (RR: 0.3, 95%CI, 0.1 - 0.5).
Caveats
The review authors rated the quality of evidence “moderate” mostly based on a lack of blinding, inevitable based on the nature of NIPPV. They also state, however, this is “unlikely to have affected primary outcomes.”The efficacy of NIPPV is dependent on its being tolerated, and for every 9 patients treated, one was unable to tolerate NIPPV. Therefore, successful treatment requires a conscious, cooperative patient. Another consideration is difficulty accessing airways due to the mask. This could limit suctioning secretions and result in aspiration or atelectasis, though these data suggest such complications were not more common with NIPPV.1
Since publication of this review the European Respiratory Society/American Thoracic Society has published practice guidelines for NIPPV in patients with COPD during acute exacerbation.2 These guidelines advise against NIPPV to prevent respiratory acidosis but recommend its use to treat acute respiratory acidosis (pH < 7.35). They also recommend NIPPV for those with severe acidosis and severe distress, as an alternative to invasive ventilation.
In conclusion, NIPPV reduces mortality and endotracheal intubation in hypercapnic respiratory failure due to COPD exacerbation. Inability to comply with treatment is higher with NIPPV, however, complications unrelated to NIPPV are less common, and complications related to NIPPV are typically minor. Because of major benefits and absence of serious harms we have assigned a color recommendation of Green (Benefit > Harm) for this intervention.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
See theNNT.com's previous reviews of this topic:
Non-Invasive Positive Pressure Ventilation for COPD Exacerbation, February 20, 2013
Author
Bryan Zorko, MD; Michael Ritchie, MDSupervising Editors: Kabir Yadav, MD; Allan Wolfson, MD
Published/Updated
September 3, 2019References:
Utility of Spinal Immobilization in Patients with Penetrating Trauma
Benefits in NNT
No one benefitted
Harms in NNT
10
1 in 10 were harmed (died)
View As:
Source
Velopulos CG, Shihab HM, Lottenberg L, Feinman M, Raja A, Salomone J, et al. Prehospital spine immobilization/spinal motion restriction in penetrating trauma: A practice management guideline from the Eastern Association for the Surgery of Trauma (EAST). J Trauma Acute Care Surg. 2018;84:736-744.Study Population: 24 studies comprising 155,089 total patients.
Efficacy Endpoints
Mitigation of neurologic deficit and potentially reversible deficit.Harm Endpoints
MortalityNarrative
Spinal precautions are a key component of many emergency medical services (EMS) protocols.1, 2 However, there is limited evidence regarding the ability of spinal immobilization (i.e. cervical collars and/or longboards) to improve patient outcomes among those with penetrating trauma, and spinal immobilization may increase complications.3, 4 These complications include increased intracranial pressure, local pressure injury, missed penetrating injury, and delay in the successful performance of vital procedures (e.g. endotracheal intubation).1, 3, 4 Moreover, even if a cervical spine collar or longboard is properly applied, patients are often not adequately immobilized.1 While prior evidence suggested that few EMS and emergency department (ED) providers were aware of the potential harms with spinal immobilization in penetrating trauma,5 it has not been established that this potentially harmful intervention actually improves patient-relevant outcomes.Investigators for the Eastern Association for the Surgery of Trauma (EAST) conducted a systematic review and meta-analysis which included randomized controlled trials, prospective observational or retrospective studies, and case-control studies evaluating the effects of spinal immobilization in adults with penetrating trauma (gunshot or stab wounds).1 Patients >13 years were considered to be adults, as these patients are typically treated as adults in many centers. Spinal immobilization was defined as the use of a cervical collar and/or longboard. The primary outcomes were mortality, neurologic deficits, and potentially reversible neurologic deficits (defined as deficit that could be either improved or reversed with definitive spinal immobilization). Secondary outcomes included missed injury and failed intubation. If pooling of data was inappropriate (moderate to high heterogeneity), the authors conducted a qualitative instead of quantitative analysis.
The systematic review included studies (n=155,089) that met the inclusion criteria for qualitative analysis and five studies (n=46,092) were suitable for quantitative analysis.1 All included studies were retrospective. No study demonstrated a benefit of spinal immobilization for mortality and neurologic injury. The incidence of neurologic injury was low, ranging from 2 to 76 per 1,000 patients. Studies focusing on patients with head and neck injuries found a higher incidence of neurologic injury, with 136 to 204 per 1,000 patients. Rates of potentially reversible neurologic injury were consistently very low, as well. Quantitative analysis (meta-analysis) of the five appropriate studies found an increased risk of harm with regard to mortality (Relative Risk [RR]: 2.4, 95% confidence interval [CI], 1.07 to 5.4; absolute risk difference [ARD]: 10.1%, 95% CI, 0.5% to 31.7%; and number needed to harm [NNH]: 10). There was no statistically significant difference for neurologic deficit (RR: 4.16, 95% CI, 0.56 to 30.89) or potentially reversible deficit (RR: 1.19, 95% CI, 0.83 to 1.70), although the point estimates favored no immobilization. There were insufficient data to perform quantitative analysis regarding failed intubation or missed injury.
Caveats
While this meta-analysis suggests that spinal immobilization in penetrating trauma is associated with increased mortality and does not reduce the risk of neurologic injuries, several limitations should be noted. All the included studies were retrospective and thus subject to the limitations inherent in this study design. The majority of studies assumed that spinal immobilization was performed based on protocol, but few studies described the type or extent of immobilization. Many studies evaluated only the projected risk versus benefit through assessment of the presence of true injury. The studies varied in their definition of the “potential benefit” of spinal immobilization, especially in regard to potentially preventable neurologic deficits. Additionally, the meta-analysis did not analyze penetrating head injury and penetrating neck injury separately. Some studies utilized surgical fixation as a surrogate outcome for reversible neurologic deficit, but these studies found that fixation may have prevented worsening of injury that had already occurred, rather than reversing it.Only five studies were designated for quantitative analysis. For mortality, the pooled estimate relied heavily on two studies,6, 7 one of which (n=45,284 patients) contributed most of the events.7 Moreover, a disproportionate number of patients were in the no-immobilization group versus the immobilization group. While the data suggest a number-needed-to-harm of 10, this may be related to bias in the single large retrospective study comprising the majority of the included patients.7 For mortality, the risk of bias was judged to below, and the quality of evidence moderate. For potentially reversible neurologic deficit, the risk of bias was low but the included studies varied widely in the definition of “potentially reversible”, which, given the rarity of injury, resulted in imprecision and wide CI’s.
Nevertheless, this analysis suggests that spinal immobilization in adults with penetrating trauma is associated with an increase in mortality and not only no benefit, but apparent actual harm in terms of neurologic deficit or potentially reversible neurologic deficits. We have thus assigned a color recommendation of Red (Harm > Benefits). Spinal immobilization is not recommended for routine use in penetrating trauma.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Brit Long, MD; Alex Koyfman, MD; Michael Gottlieb, MD, RDMSSupervising Editors: Joshua Quaas, MD; Allan Wolfson, MD
Published/Updated
References:
Outpatient Treatment for Low-Risk Febrile Neutropenia
Benefits in NNT
NA (No difference in risk of treatment failure or mortality in adults or children)
Shorter hospitalization by 1.64 days in adult patients and 3.9 days in pediatric patients
Harms in NNT
No difference in adverse drug reactions
View As:
Source
Rivas-Ruiz R, Villasis-Keever M, Miranda-Novales G, Castelán-Martínez OD, Rivas- Contreras. Outpatient treatment for people with cancer who develop a low-risk febrile neutropaenic event. Cochrane Database Syst Rev. 2019 Mar 19;3:CD009031.Study Population: 10 trials including 994 low risk patients (628 adults, 366 children) with cancer, fever, and neutropenia.
Efficacy Endpoints
Treatment failure, mortalityHarm Endpoints
Adverse drug reactionsNarrative
Fever and infection are common in neutropenic cancer patients.1,2 While some become severely ill, most patients have an uneventful course, with 50-60% having no life-threatening complication or fatal infection.1,2 Patients with febrile neutropenia have therefore been divided into low-risk and high-risk groups. Those patients at low-risk of complications may benefit from outpatient management.1,2 Admission to the hospital has its own risks, including iatrogenic infections and reduced quality of life.1 Guidelines thus recommend risk stratification for potential outpatient treatment.3,4 However, it is important to determine if outpatient management is as safe and effective as inpatient management in low-risk patients.This systematic review and meta-analysis5 included randomized controlled trials that compared inpatient antimicrobial therapy with outpatient antimicrobial therapy for low-risk febrile neutropenic adults or children with cancer. The primary outcomes were treatment failure (death, non-resolution of signs or symptoms of presenting infection, or change of antibiotic) and mortality at 30 days.
The authors identified 10 relevant studies (n=994), six in adults (n=628) and four in children (n=366). Definitions for low risk were not standardized, but generally required that patients not a) need hospitalization, b) have focal or severe infection, c) have relapse of the disease, and d) be receiving intensive chemotherapy. Overall, there were no difference in treatment failure (relative risk [RR] 0.81, 95% confidence interval [CI] 0.32 to 2.71) or mortality (RR 1.11, 95% CI 0.41 to 3.05). Among adults there was no difference in treatment failure (RR 1.2, 95% CI 0.8 to 1.9) or mortality (RR 1.0, 95% CI 0.3 to 3.7). Among pediatric patients, there was also no difference in treatment failure (RR 1.0, 95% CI 0.6 to 2.0) or mortality (RR 0.6, 95% CI 0.2 to 2.7). Hospitalization duration, a secondary outcome, was 1.64 days lower in the adult outpatient group (95% CI -2.22 to 1.06) and 3.9 days lower in the pediatric outpatient group (95% CI 95% CI -5.37 to - 2.43). The risk of adverse drug reactions (harm endpoint) was not statistically significant between the two groups (low quality evidence).
Caveats
While this review suggests no significant difference in treatment failure or mortality between inpatient and outpatient management, patients were observed for 24 to 72 hours in the hospital before discharge in 6 trials and discharged immediately in only 2 trials. Despite this, there was still a reduction in patient hospitalization and length of stay. The certainty of this estimate was considered low, however, based on potential bias and quality of evidence.Additionally, low-risk criteria varied between studies with only one utilizing Multinational Association for Supportive Care in Cancer (MASCC) criteria and none using Clinical Index of Stable Febrile Neutropenia (CISNE) criteria, the two currently recommended tools.3,4,5 There are no existing criteria for low-risk stratification in pediatric patients with neutropenic fever. Moreover, there were differences in types and routes of antibiotic regimens and types of cancers (eg, bloodborne versus solid tumors). There was also limited reporting on randomization and allocation concealment. Evidence quality of the included RCTs was low to moderate based on the GRADE approach, and confidence intervals were wide for main outcome measures. Finally, the studies may have been underpowered for their primary outcomes due to low sample sizes in several trials.
Despite the above limitations, these findings suggest outpatient treatment of selected low-risk patients with cancer and febrile neutropenia was, in these investigations, as safe as inpatient management. We have assigned a color recommendation of Yellow (Unclear if Benefits) both because the only quantifiable benefit was a secondary measure, and because of the low certainty of this finding. Clearly, larger, high quality trials are needed to establish with more certainty the promising benefits suggested by these data.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Michael Gottlieb, MD; Alex Koyfman, MD; Brit Long, MDSupervising Editor: Michael Ritchie, MD; Dan Runde, MD
Published/Updated
August 1, 2019References:
Tranexamic Acid for Postpartum Hemorrhage
Benefits in NNT
No one was helped (no death was prevented)
Harms in NNT
No one was harmed
View As:
Source
Effect of early tranexamic acid administration on mortality, hysterectomy, and other morbidities in women with post-partum haemorrhage (WOMAN): an international, randomised, double-blind, placebo-controlled trial. WOMAN Trial Collaborators. Lancet. 2017; 389: 2105–2116.Study Population: 20,060 women aged 16 yrs and older with clinical diagnosis of postpartum hemorrhage after vaginal birth or cesarean section from 1 randomized control trial
Efficacy Endpoints
Death from all causes, death from bleeding, and hysterectomyHarm Endpoints
Thromboembolic events and organ failureNarrative
Postpartum hemorrhage (PPH) is the most common cause of maternal death worldwide. Tranexamic acid (TXA) is an antifibrinolytic agent that has shown to decrease bleeding in surgical patients and all cause death in trauma patients.1, 2 In 2012, TXA was incorporated into the World Health Organization (WHO) guidelines for refractory or trauma-related PPH.3The WOMAN trial4 was a single randomized, double-blind, placebo controlled, multi-center international study consisting of 20,060 women age 16 years and older with a diagnosis of PPH after vaginal birth or C-section. It investigated whether or not early administration of TXA reduced the rate of death and hysterectomy in patients with PPH when compared to placebo. One comparison was made -- TXA versus placebo. Outcomes were measured at hospital discharge or on day 42 if still in the hospital.
Benefits: Administration of tranexamic did not reduce all-cause mortality. However, TXA did show a reduction in the risk of death due to bleeding (Relative Risk [RR]: 0.81, 95% CI 0.62 to 0.98; Absolute Risk Difference [ARD]: 0.4%; NNT 267, very low quality evidence). There was no difference in hysterectomy rate between the two groups.
Harms: There was no difference between TXA and placebo in the rate of thromboembolic events (deep vein thrombosis, pulmonary embolism, myocardial infarction and stroke), organ failure (renal, cardiac, respiratory) or sepsis and use of uterotonics.
A recent systematic review and meta-analysis5 which only included postpartum hemorrhage following vaginal delivery and excluded cesarean sections (approximately 14000 patients from two RCTs, including the subset of patients from WOMAN trial who had postpartum hemorrhage after vaginal delivery) similarly showed no all-cause mortality benefit. This meta-analysis however showed reduced risk of hysterectomy (RR: 0.63, 95% CI, 0.42–0.94; ARD: 0.3%; NNT: 333, very low quality evidence) after postpartum hemorrhage resulting from vaginal delivery. This analysis also showed the tranexamic acid did not increase the risk of thromboembolic events, stroke, heart attack, or sepsis.
Caveats
One caveat is that the subjective inclusion criteria may have introduced selection bias into the study. Clinical diagnosis of PPH was based on the ability to estimate blood loss (500 mL after vaginal delivery or 1,000 mL C-section) which can vary between clinicians. Hemodynamic instability was also an inclusion criteria but objective measurements to define this were not discussed. Additionally, patients were enrolled only if clinicians were uncertain about giving TXA leaving out subgroup of patients with more severe pathology who were given TXA as standard of care. Excluding these patients, especially those with hemodynamic instability from severe hemorrhage may have lead to an underestimation of the TXA’s true efficacy.Another caveat to consider is that the sample size was significantly increased from 15,000 to 20,000 after investigators discovered that the decision to perform hysterectomy was commonly made at the time of randomization and thus could not be impacted by intervention. This runs into the risk of overpowering which can interpret seemingly small and unremarkable differences between treatment and control arms as statistically significant.
Although we only reported individual outcomes in our review, the original study selected a composite outcome (death from all-causes or hysterectomy within 42 days of giving birth) as their primary outcome. The authors interpreted their results as positive despite the lack of statistical difference in this primary composite outcome. This was partly from shifting focus from all-cause mortality to cause-specific mortality specifically death due to bleeding. Although there was significantly less death from bleeding in the TXA group, the calculated fragility index was zero pointing to the lack of robustness of this dataset. We also do not consider disease-specific mortality a true efficacy endpoint.
In light of the results of this trial, WHO recommends administration of tranexamic acid to all patients with postpartum hemorrhage.3 This recommendation is not supported by the existing evidence with regards to all-cause mortality. Therefore we have assigned an NNT color recommendation of yellow (Unclear if Benefits). However, considering the safety and low cost of tranexamic acid ($45-$55 per vial), and the potentially devastating consequences of postpartum hemorrhage, we believe TXA should be considered as an attractive adjunctive therapy when other modalities (e.g. treating uterine atony) fail to control the hemorrhage.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Eric Tang, MD; Jessica Stetz, MDSupervising Editors: Fredrik Amell, MD; Jarone Lee, MDPublished/Updated
References:
Tranexamic Acid for Upper Gastrointestinal Bleeding
Benefits in NNT
30
1 in 30 were helped (death prevented) when compared to placebo; no one was helped when compared against antiulcer therapy
Harms in NNT
No one was harmed
View As:
Source
Bennett C, Klingenberg SL, Langholz E, Gluud LL. Tranexamic acid for upper gastrointestinal bleeding. Cochrane Database of Systematic Reviews 2014, Issue11. Art. No.:CD006640. DOI: 10.1002/14651858.CD006640.pub3.Study Population: 1701 patients from 8 randomized controlled trials with acute upper gastrointestinal bleeding
Efficacy Endpoints
Death, re-bleeding, and requirement for surgeryHarm Endpoints
Thromboembolic events, myocardial infarction, pulmonary embolism, cerebral infarction, or deep vein thrombosisNarrative
Upper gastrointestinal bleeding is common and accounts for at least half of the nearly 500,000 annual U.S. hospitalizations for gastrointestinal bleeding.1 In the acute setting, severe bleeding is treated with intravenous fluids, blood products, antiulcer therapy, and hemostasis by endoscopy.2 Tranexamic acid (TXA) is an antifibrinolytic agent shown to reduce bleeding.3, 4 TXA has been proven to be effective in improving patient-centered outcomes after severe hemorrhage due to trauma.5 The authors of this systematic review sought to evaluate the benefit of TXA administration specifically for upper gastrointestinal bleeding.The systematic review summarized here6 identified 8 randomized trials of TXA in 1701 subjects presenting with acute upper gastrointestinal bleeding among patients admitted to the hospital, including some in the intensive care unit. Two comparisons were made: TXA versus placebo and TXA versus antiulcer therapy (cimetidine or lansoprazole). Primary outcomes were mortality and adverse events. Compared to placebo, TXA reduced mortality (relative risk [RR]: 0.60, 95% CI 0.42 to 0.87; ARR: 3.5%; NNT: 30, moderate quality evidence). However, because of a high attrition in several trials the results must be interpreted with caution. About 20% of the studied patients were withdrawn or excluded for reasons such as lack of confirmation of the presence of bleeding, presence of malignancy, terminal illness, or late administration of treatments. Re-analysis including all participants and considering missing patients as treatment failures did not show mortality benefit.5
In the second comparison, TXA versus antiulcer therapy (cimetidine or lansoprazole), only two trials were included, and no mortality benefit was found.
Administration of TXA did not reduce the risk of re-bleeding (RR 0.72, 95% CI 0.50 to 1.03, low quality evidence:) or blood transfusion (RR 1.02, 95% CI 0.94 to 1.11, very low quality evidence:).
Although meta-analysis could not be performed for harm endpoints due to lack of adverse event reporting for all trials, three studies did include data on thromboembolic events. There was no difference between the TXA and placebo groups in combined serious thromboembolic events (myocardial infarction, pulmonary embolism, and cerebral infarction (RR: 1.37, 95% CI 0.36 to 5.28), nor did TXA increase the risk of deep vein thrombosis (RR: 2.32, 95% CI 0.60 to 8.89).
Caveats
The authors of this Cochrane review judged the available evidence to be of moderate to low quality, largely due to the risk of bias and clinical heterogeneity among included trials. Notably, the trials were conducted over nearly four decades (from 1973 to 2011), with 6 of 8 published between 1973 and 1987, likely accounting for much of the heterogeneity. A high drop-out rate was also concerning. When this was accounted for (in a worst-case scenario), the mortality benefit was not significant. The included trials also used different doses and routes of administration for TXA, and were mostly performed 30-45 years ago. Management patterns, hemostatic technology, and co-interventions have since changed, in some cases dramatically, making applicability to current practice questionable. Lastly, all trials enrolled admitted patients. Previous trials have shown that TXA is most efficacious when administered early (within one hour).5 Therefore, the delay in administration of TXA might have reduced efficacy, further reducing applicability and generalizability for emergency department patients.We have assigned a color recommendation of Yellow (unclear benefits) to this intervention. Limitations of the reported data, particularly the lost to follow-up and dropout rates, the high risk of bias, and the presence of significant heterogeneity justify this rating. A large pragmatic double-blind controlled trial with a target sample size of 12,000 subjects is currently ongoing.7 We are hopeful this trial will provide better evidence. Despite TXA’s lack of demonstrated benefit compared to standard treatments with respect to the endpoints of mortality or re-bleeding, given the relative safety, lack of significant adverse events, and low cost of the medication, it may be reasonable to consider TXA in severe upper gastrointestinal bleeding as an adjunct to standard therapy, or if standard therapy fails.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Raymond Beyda, MD; Davood Johari, MDSupervising Editors: Kabir Yadav, MD; Shahriar Zehtabchi, MDPublished/Updated
July 1, 2019References:
Branched-Chain Amino Acids for People with Hepatic Encephalopathy
Benefits in NNT
6
1 in 6 had improvement in signs and symptoms of hepatic encephalopathy assessed mainly by West haven criteria
Harms in NNT
20
1 in 20 were harmed by adverse effects like nausea and diarrhea
View As:
Source
Gluud LL, Dam G, Les I, et al. Branched-chain amino acids for people with hepatic encephalopathy. Cochrane Database Syst Rev. 2017;5:CD001939. doi: 10.1002/14651858.CD001939.pub4Study Population: Adults with hepatic encephalopathy and cirrhosis, mostly due to alcoholic liver disease or viral hepatitis
Efficacy Endpoints
Mortality, Hepatic encephalopathyHarm Endpoints
Gastrointestinal symptoms, including nausea and diarrhea; and any other reported adverse effectsNarrative
Cirrhosis, where scar tissue replaces normal hepatic tissue, is the most common cause of hepatic encephalopathy,1 a brain dysfunction that can be mild with minimal confusion, or overt and severe with coma.1, 2, 3 Branched-chain amino acids (BCAAs) may reduce hepatic encephalopathy by helping skeletal muscle detoxify blood4, 5, 6 and are therefore recommended by some as routine treatment.2, 3The systematic review summarized here identified 16 randomized trials of 827 subjects (97% with cirrhosis) with hepatic encephalopathy.1 Primary outcomes were mortality, hepatic encephalopathy (number with improvement), and harms. Subjects were followed for varying time periods ranging from 1 to 104 weeks.
BCAAs had benefits in improving signs and symptoms of hepatic encephalopathy assessed mainly by West haven criteria (RR 0.7, 95% CI 0.6 to 0.9 ARD: 17%, NNT: 6): for every 6 subjects treated, one measurably improved. However, no effect was found on mortality (RR 0.9, 95 % CI 0.7 to 1.1). BCAAs also increased nausea and diarrhea (RR 3.4, 95% CI,0.7 to 16.5, ARD: 5%, NNH: 20); though, no serious adverse events were reported.
The West-Haven criteria classifies the degree of mental status disturbance in encephalopathy by 4-point scoring system ranging from reversal of sleep patterns and mild alteration in cognition to deep coma.2
The portal-systemic encephalopathy (PSE) index may also objectively describe the overall clinical severity of HE.7 It is calculated following assessment of five elements; Mental status (Evaluated by West-Haven criteria), presence and intensity of asterixis, time taken to complete psychometric tests of intellectual function (such as number connection test), venous ammonia level and electroencephalogram (EEG) abnormalities.7
In this metanalysis, Six studies (Cerra 1985, Hwang 1988, Muto 2005, Strauss 1986, Vilstrup 1990, Rossi-Fanelli 1986) assessed improvement of hepatic encephalopathy strictly only by West haven scoring only.1 However, the other 10 studies (Fiaccadori 1984, Horst 1984, Michel 1985, Egberts 1985, Calvey 1985, Marchesini 1990, Hayashi 1991, Plauth 1993, Les 2011, Marchesini 2003) not only included West haven criteria to evaluate degree of encephalopathy, but they also included some or all components of PSE Index.1
Caveats
The authors of the Cochrane review graded the evidence as high quality for hepatic encephalopathy. However, there was moderate heterogeneity of their results, which raises concerns about validity and applicability. In addition, most trials were non-blinded, small, and judged to be at “high risk of bias.” The ‘high quality’ grade here seems highly debatable; and a large, well-done clinical trial may easily upend these findings.The review also pooled trials using different forms of BCAA.1 Nine assessed oral and seven assessed intravenous administration, with only oral showing a statistical benefit. In addition, assessing hepatic encephalopathy is fraught with subjectivity and disagreement. For instance, many studies in this review used the PSE index, which the Food and Drug Administration has rejected as inadequate.8 This is mainly due to the inclusion of blood ammonia levels and severity of asterixis.8 The utility of ammonia level is controversial given that ammonia concentration is not useful for screening for hepatic encephalopathy since their levels vary if they are arterial or venous.9, 10 In addition too, due to the fact that these levels are significantly affected by collection techniques and can be falsely elevated if the sample was collected after fist clenching, using tourniquet, or if the sample was not placed on ice.10
Also, asterixis is not specific to HE as it can also be observed in patients with other forms of metabolic encephalopathies such as in uremia and respiratory failure.10
Without blinded assessors, a method not used in most trials here, and a validated scale, it is difficult to have confidence in these findings. This is particularly true when the only objective outcome, mortality, showed no difference between groups.
The yellow color recommendation (unclear benefits) is based on the inconclusive data supporting benefits of BCAA for hepatic encephalopathy.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Ahmed Hamed MD; Amira Hamed MD; Karissa Lambert MDSupervising Editors: Michael Ritchie, MD; James McCormack, MD
Published/Updated
June 14, 2019References:
High-flow Oxygen Therapy for Treating Bronchiolitis in Infants
Benefits in NNT
9
9 for preventing escalation of care
Harms in NNT
No difference between the groups for adverse events
View As:
Source
Franklin D, Babl FE, Schlapbach LJ, et al. A randomized trial of high-flow oxygen therapy in infants with bronchiolitis. N Engl J Med 2018;378:1121–31.Study Population: 1,472 infants younger than 12 months with signs of bronchiolitis with oxygen requirement
Efficacy Endpoints
Treatment failure (requiring escalation of care), admission to intensive care unit, duration of hospital stay, the duration of intensive care unit stay, duration of oxygen therapy, intubation ratesHarm Endpoints
Serious adverse events including pneumothorax, respiratory arrest, cardiac arrest, apnea, emergency intubationNarrative
Bronchiolitis is the most common reason for hospitalization in infants worldwide.1 Current recommendations by the American Academy of Pediatrics are for supportive care including maintenance of hydration and oxygen support for hypoxemia.1 Other interventions such as the use of bronchodilators have failed to show any benefit when compared to supportive care alone. However, it has been proposed that the obstructive process of bronchiolitis that causes increased work of breathing, hypoxia, and hypercapnea might respond to the moderate positive pressure provided by high-flow oxygen therapy.2The randomized control trial referenced here was conducted in Australia and New Zealand across multiple institutions on otherwise healthy infants (less than 12 months old) with bronchiolitis with an oxygen requirement.3 For the purposes of the study, oxygen requirement was defined as the need for supplemental oxygen to maintain oxygen levels between 92% and 98% (11 institutions used site-specific standard of 94%–98%).3 Patients were randomized to heated and humidified high-flow oxygen at a rate of 2 L/kilogram body weight/min delivered by the Optiflow system with the use of an age-appropriate Optiflow Junior cannula and the Airvo 2 high-flow system (intervention group) or supplemental oxygen through a nasal cannula, up to a maximum of 2 L/min, to maintain an oxygen-saturation level in the range of 92% to 98% (control group).3
Treatment failure was defined as the need for escalation of care based on standardized clinical criteria: persistent or worsening tachycardia, tachypnea, worsening of hypoxemia requiring >40% FiO2 in the high-flow oxygen group and >2 L/min flow rate of nasal cannula in the standard therapy group. Each hospital was allowed to use its own escalation protocol to be used as the criteria for treatment failure. Each episode of escalation of care was reviewed to ensure that it met study criteria. Escalation of care in the standard oxygen group was recommended to switch each patient to high-flow therapy.
The trial showed a 11% absolute risk reduction in the need for escalation of care in patients receiving high-flow oxygen therapy (relative risk = 0.52, 95% confidence interval = 0.40–0.66; NNT = 9). This trial did not show any significant difference between the groups for other outcomes such as duration of hospital or ICU stay and intubation rates (although a very small percentage of patients [12/1,472] required intubation).3
High-flow oxygen therapy did not result in any significant increase in the risk of adverse events, although the rate of adverse events was very low in both groups and no patients in any of the groups required emergency intubation and cardiopulmonary resuscitation. One child in each group was diagnosed with pneumothorax but none required thoracostomy.3
Similar results were found by another recent smaller trial that reported an absolute risk reduction of 9% in treatment failure rate (NNT = 11) in patients allocated to high-flow oxygen therapy but no statistically significant difference between the groups for time to oxygen weaning or length of stay. The rates of adverse events were similar between the two groups in this trial as well.4
Caveats
This is the largest randomized trial to date addressing this important research question.3 The major limitation of this trial was the absence of blinding, which was not possible due to difference between the equipment. To reduce the risk of bias, the investigators remained blinded to the trial outcome until the trial was completed.The primary outcome of this trial was treatment failure defined as requiring escalation of care. This was a composite outcome which reflected admission to a higher level of care or changing from low-flow oxygen to high-flow oxygen therapy (control group) and may not be considered a patient-centered outcome. In addition, determining this outcome was somewhat subjective. Analyzing individual patient-centered outcomes such as length of hospital or ICU stay and intubation rate did not show any benefits from using high-flow oxygen therapy. It must be noted that according to the Australian New Zealand Clinical Trials Registry, the initial primary outcome of the trial was reduction in transfer rate from regional hospital to tertiary center. This outcome was changed after inclusion of tertiary centers since this outcome would not be applicable anymore for patients who present directly to a tertiary emergency department (ED).5
While the overall rate of treatment failure and the need for escalation of care was lower in patients allocated to high-flow oxygen therapy, when the high-flow group was divided by hospital with an on-site pediatric intensive care unit (PICU) versus no PICU, the escalation rate was significantly higher in hospitals with an on-site PICU (14% vs. 7%). Therefore, availability of an on-site PICU could be an important factor in escalation of care by treating physicians.3
It is notable to mention that 61% of the patients in the standard therapy group who experienced treatment failure were transitioned to high-flow oxygen therapy and responded positively.3 High-flow oxygen therapy may potentially have the highest overall benefit in hospitals without an intensive care unit as it may decrease the need for interfacility transfers.
Another limitation of the reported data is that 34% of all patients that had escalation of care did not meet the criteria for escalation of care based on the study criteria but met the individual hospitals escalation criteria. This can present some confounding when looking at treatment failure between the groups.3
It must be noted that the trial did not control for the effect of high-flow oxygen therapy itself as a main factor for the need for higher level of care. Assignment to high-flow oxygens above 2 L/kg might have prompted certain physicians to escalate the level of care for closer observation and higher demands for nursing care.
The trial discussed in this review did not exclusively enroll patients in the ED.3 Patient enrollment occurred both in the ED and on the pediatric wards. Therefore, a trial originated exclusively in the ED might produce different results. Er et al.6 explored the characteristics of ED patients with bronchiolitis who respond poorly to high-flow oxygen therapy. These investigators concluded that low initial oxygen saturation, respiratory acidosis, and an oxygen saturation/ fraction of inspired oxygen ratio less than 195 at the first hours of treatment were related to unresponsiveness to high-flow oxygen therapy in the pediatric ED.5
Unfortunately, this trial does not evaluate the cost effectiveness of high-flow oxygen therapy. Other published trials have suggested cost saving benefits from using high-flow oxygen therapy.4, 7, 8, 9 Kepreotes et al.4 discussed the estimated cost savings with the use of high-flow oxygen therapy and concluded that high-flow oxygen therapy might have a role as a rescue therapy to reduce the proportion of children requiring high cost intensive care. Heikkilä et al.,7 performed a cost analysis of high-flow oxygen therapy versus standard oxygen therapy and found that using high-flow oxygen therapy was associated with a $441 saving per patient due to decreases in ICU admission and hospital transfers. Finally, this trial used pulse oximetry levels of 92% to 98% (94%–98% in specific institutions) to evaluate response to therapy while the American Academy of Pediatrics recommends initiation of oxygen therapy at pulse oximetry levels of 90% or below.10
In conclusion, high-flow oxygen therapy in infants with bronchiolitis reduces the risk of treatment failure and the need for escalation of care. However, it does not offer any benefit as far as direct patient-centered outcomes are concerned. Therefore, we assign a color recommendation of yellow (unclear benefits) to this intervention. However, this trial still has clinical implications. It appears that for patients with bronchiolitis who do not respond to low-flow oxygen therapy (first line of therapy) based on criteria used in this trial or other institutional criteria, high-flow oxygen therapy should be considered as the next logical step before employing other more aggressive measures.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Isaac Gordon, MD; Ambreen S. Khan, MDSupervising Editor: Kabir Yadav, MD
Published/Updated
References:
Aspirin For Preventing A First Heart Attack Or Stroke
Benefits in NNT
No deaths were prevented
333
1 in 333 avoided a nonfatal heart attack
Unclear if ischemic strokes avoided
Harms in NNT
250
1 in 250 suffered a major bleeding event
View As:
Source
Bibbins-Domingo K. Aspirin Use for the Primary Prevention of Cardiovascular Disease and Colorectal Cancer: U.S. Preventative Service Task Force Recommendation Statement. Ann Intern Med. 2016;164:836-845.Mahmoud AN, Gad MM, Elgendy AY, Elgendy IY, Bavry AA. Efficacy and safety of aspirin for primary prevention of cardiovascular events: a meta -analysis and trial sequential analysis of randomized controlled trials. Eur Heart J. 2019;40:607 -17.
Zheng SL, Roddick AJ. Association of Aspirin Use for Primary Prevention With Cardiovascular Events and Bleeding Events: A Systematic Review and Meta-analysis. JAMA. 2019;321:277–87.
Study Population: Approximately 164,000 subjects at varying risk for cardiovascular disease.
Efficacy Endpoints
Death, heart attack, stroke, measured over 5-7 years.Harm Endpoints
Major bleeding events, hemorrhagic strokes.Narrative
Cardiovascular disease (CVD) is a major cause of death worldwide. Aspirin inhibits platelet aggregation which reduces clot formation. Thus, aspirin can help prevent cardiovascular problems caused by blood clots, but it can also increase risk of bleeding. For persons with known CVD, the beneficial effect of aspirin use for preventing cardiovascular events outweighs the harmful side effects (e.g. bleeding).1 The efficacy of aspirin in preventing cardiovascular events in patients without previously known CVD (primary prevention), has been unclear. This evidence-based summary examines the benefits and harms of aspirin in patients without known CVD. For this purpose, we summarize the 2015 US PreventiveServices Task Force report2 and two recent systematic reviews of aspirin for primary prevention.3, 4 The USPSTF report, published in 2016, was the definitive systematic review until three trials were published after its release. The ARRIVE,5 ASCEND,6 and ASPREE7 clinical trials together included over 47,000 new subjects. Two updated systematic reviews by Mahmoud et al.3 and Zheng et al.4 were published in 2019 and both include all the most recent trials. The summary table above of benefits and harms are derived from the two most recent meta-analyses by Mahmoud et al.3 and Zheng et al.4 because the USPTF report does not include the most recent trials. For the most part, the reported number-needed-to-treat (NNT) values and number-needed-to-harm (NNH) values were similar between the two reviews (Table 2). When the NNT and NNH values were different, to be conservative, we reported the higher NNT value and the lower NNH value.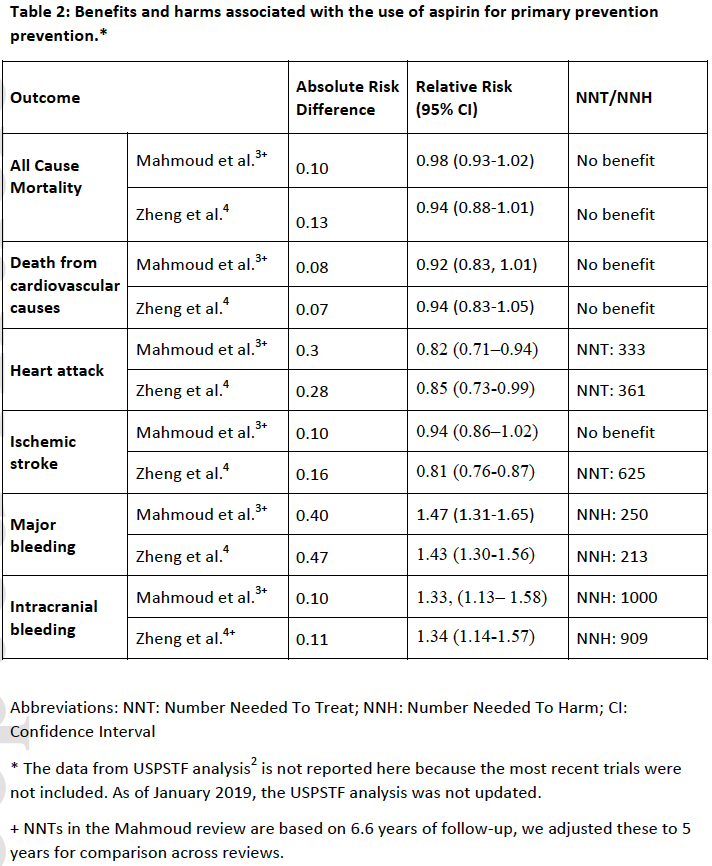
Mortality:
For the outcome of greatest interest, mortality, the older USPSTF analysis found a statistically significant overall benefit for aspirin at all doses, but no statistically significant benefit for aspirin at low doses (100 mg or less). The two updated reviews by Mahmoud et al.3 and Zheng et al.4 found no benefit in mortality regardless of the dose of aspirin (Table 2).
Nonfatal heart attacks:
All three reviews found statistically significant relative reductions of 15-20% in nonfatal heart attacks from the use of aspirin, with number-needed-to-treat (NNT)* values of 250,2 3333 and 361.4
Stroke:
The older USPTF analysis and the newer review by Mahmoud et al. did not find that aspirin prevented ischemic stroke, combined fatal and nonfatal.2, 3 Only the systematic review by Zheng et al.4 showed a small reduction in risk of ischemic stroke in patients allocated to the aspirin group, with an NNT value of 625 (Table 2).
Major Bleeding:
All three reviews found statistically significant relative increases of 30-50% in major bleeding events with number needed to harm (NNH) values of 142-357 (stratified by baseline risk),2 250,3 and 213.4 Major bleeding was defined differently within each trial, and could have included intracranial hemorrhage, major gastrointestinal bleeding, ocular bleeding, major epistaxis, or any extra-cranial bleeding requiring transfusion or hospitalization
Subgroups:
Harms outweighed benefits in all three reviews when analyzing all patients, with no mortality reduction. The USPSTF,2 however, projected one subgroup that may have benefits outweighing harms. USPSTF looked at different age groups and divided each age group into different CVD risk groups using the AHA calculator to predict the 10-year risk of a cardiovascular event. In a computer model by Health Partners Institute,8 50-59 year old patients with >10% risk over 10 years saw a projected benefit from reduction in nonfatal heart attacks that outpaced the increase risk of major bleeding. No other subgroup realized anet overall benefit.
Dosing:
Optimal dosing of aspirin is unknown and some data suggest weight-based dosing.9 In the USPSTF analysis, low-dose aspirin (100 mg or less) was not associated with lower mortality but higher doses were, while the opposite association was true for nonfatal stroke. Aspirin dosing was not addressed in the Mahmoud et al.3 and Zheng et al.4 reviews. The conflicting results for dose finding in the older report and lack of newer analysis in the updated reviews makes dosing risk-benefit analysis unclear at this time.
Caveats
The older USPSTF report has limitations. The finding of overall benefit for 50-59 year old patients is a computer projection based upon a statistical model.10 The model uses data from subgroups across several trials, and applies the benefits found with aspirin to a hypothetical person - in this case, a 50-59 year-old American male - with a baseline cardiovascular risk estimated using the AHA risk calculator. Unfortunately, that calculator substantially overestimates risk (by anywhere from 20-100% or more).11,s 12, 13 Given the razor-thin benefit margins found, any overestimate of baseline risk would convert the finding of overall benefit to a finding of overall harm. Moreover, the model is out of date as three new large randomized controlled trials have been published since its release.There is debate across reviews about the definition of“primary prevention.” In the ETDRS study14 half of the patients had known CVD, and all patients in the POPADAD study15 and the AAA study16 had arterial disease. The USPSTF and older meta-analyses included these studies, Mahmoud et al.3 excluded them, and Zheng et al.4 included ETDRS only. These patients constitute less than 9000 subjects (5%) of the total patients analyzed. The varying inclusion of the three studies who enrolled patients with apparent CVD resulted in 96% overlap between the Mahmoud et al. review (157000 subjects) and Zheng et al. review (164000 subjects). The inclusion of some patients with CVD in the Zheng et al. study may explain why Zheng et al. found a statistically significant small stroke prevention benefit, while Mahmoud et al. did not (Table 2).
Less clear are differences in the two new meta-analyses regarding the heterogeneity of the trials. Zheng et al. found no heterogeneity (I2=0%) for heart attack reduction, while Mahmoud et al. concluded “a high degree” of heterogeneity (I2=67%). These differences in the results could be from existing heterogeneity among different trials that were included (11 trials in the analysis by Mahmoud et al. and 13 trials in the analysis by Zheng et al.), but more likely reflects differences in the methodology of the meta-analyses.
Regardless of these minor differences, both updated reviews found no consensus finding of benefits outweighing harms in patients regardless of CVD risk, contradicting the statistical model projection from the USPTF report that high-risk subgroups may benefit specifically. While overall benefit may be true in secondary prevention in high-risk patients, results from these primary prevention reviews are uniform in the benefits not outweighing the harms. In fact, the recent ASPREE trial found high-risk patients had increased harm compared to low-risk patients.7 The proportion of high-risk patients was highest among the newer studies (25-30%), and no statistically significant benefit was found in any outcome—only harms from mortality and bleeding.5, 6, 7
Further concern regarding the USPTF report reliance on the AHA calculator to project a subgroup benefit is that coronary events occurred at less than a third the predicted rate in the ASPREE trial,7 and less than half predicted in the ARRIVE,5and ASCEND6 trials. In practice, clinicians often apply the AHA calculator used in trials and estimate risk conservatively. If faulty calculators and conservative gestalt lead to overestimation of risk, and clinicians wrongly believe higher risk means greater benefit from aspirin, overall harm due to aspirin prescribing for primary prevention is probably widespread.
By the same token, overestimation means true ultra-high-risk patients (10-year risk of cardiovascular disease >30%) were potentially misdiagnosed as having pre-existing CVD and were not enrolled in primary prevention trials and may potentially benefit. Future studies should tackle this question.
Last but not least, patient preference is an important factor for making the decision regarding aspirin use for primary prevention of cardiovascular disease. Some patients may value avoiding nonfatal heart attacks or possibly avoiding ischemic strokes as being worth the increased risk of major bleeding.
We chose a Red color recommendation (No Benefit) because of consistent findings of harm outweighing benefit. We considered Black (Harmful) but recognize there may be subgroups studies that will identify patients who can benefit.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Kristopher Roach, MD; Michael Ritchie, MD; Shahriar Zehtabchi, MDSupervising Editor: Kabir Yadav, MD
Published/Updated
References:
Procainamide Versus Amiodarone for Stable Ventricular Tachycardia
Benefits in NNT
4
1 in 4 in favor of procainamide to convert one more patient with stable VT to baseline rhythm
Harms in NNT
3
1 in 3 against amiodarone for one more patient to experience major adverse cardiac event
View As:
Source
Ortiz M, Alfonso M, Arribas F, et al. Randomized comparison of intravenous procainamide vs. intravenous amiodarone for the acute treatment of tolerated wide QRS tachycardia: the PROCAMIO study. Eur Heart J 2017;38:1329–35.Study Popluation: 74 adults with hemodynamically stable, wide-QRS tachycardia.
Efficacy Endpoints
Pharmacologic conversion to baseline rhythm within 40 minutes without need for electrical cardioversion.Harm Endpoints
Major cardiac adverse events within 40 minutes of administration: clinical hypoperfusion, acute heart failure, hypotension, increased tachycardia, polymorphic ventricular tachycardia.Narrative
Electrical cardioversion is an effective treatment for termination of ventricular tachycardia (VT)1, 2 but is typically performed with procedural sedation and thus involves associated risk. In hemodynamically stable VT, pharmacologic cardioversion is an option. Historically, lidocaine, amiodarone, procainamide, and sotalol have been used for pharmacologic cardioversion, based mostly on expert opinion. Lidocaine has fallen out of favor because it was shown to be inferior to both procainamide and sotalol.1, 3, 4, 5 The use of amiodarone, initially suggested based on extrapolation from cardiac arrest treatment,6 has been challenged by two retrospective analyses, albeit limited in design.7, 8 The American Heart Association (AHA) recommends procainamide (IIa) over amiodarone (IIb) for pharmacologic conversion of VT,1, 4 whereas the European Resuscitation Council (ERC) favors amiodarone.5A prior retrospective study9 failed to demonstrate a difference in efficacy between amiodarone and procainamide for cardioversion of stable VT. The trial discussed here10 is the only known randomized controlled trial (RCT) that compares the two drugs for this purpose. This multicenter study randomized 74 patients to receive procainamide (10 mg/kg over 20 minutes) or amiodarone (5 mg/kg over 20 minutes) for stable wideQRS complex tachycardia presumed to be VT. The researchers found that subjects in the procainamide group were significantly less likely to experience harm as the primary outcome, a major adverse cardiac event (odds ratio [OR] = 0.1, 95% confidence interval [CI] = 0.04 to 0.6, absolute risk difference [ARD] = 32%, NNH = 3). They also report an advantage in a secondary outcome, conversion to baseline rhythm (OR = 3.3, 95% CI = 1.2 to 9.3, ARD = 29%, NNT = 4).
Caveats
Stable VT is uncommon in most settings and therefore difficult to investigate prospectively. This study was small with 74 subjects (out of a calculated sample size of 302) enrolled over 6 years. It was stopped early due to difficulty in enrollment.In addition to a small sample size, early stoppage, a lack of blinding, and unclear concealment of allocation, there are other potential confounding factors. Two electrophysiologists judged 90% of rhythms to be “probable/definite VT” implying that up to 10% of enrolled patients may have had supraventricular tachycardia (SVT). The prevalence of underlying cardiomyopathy and risk of hypotension may have differed in the proportions with SVT and VT. SVT can be terminated by amiodarone,11 and procainamide has been previously recommended for treatment of refractory SVT.12 Without knowing the comparative effectiveness of amiodarone and procainamide for SVT, potential contamination of 10% of patients having SVT has an unknown effect on the results.
The study excluded patients with dyspnea or anginal symptoms. Acute ischemia-related VT may respond to drugs differently than VT unrelated to acute ischemia. The trial also excluded patients whose wide-QRS tachycardia was terminated with adenosine prior to randomization. These patients were considered to have SVT; however, a proportion of idiopathic VTs will respond to adenosine therapeutically.1 The exclusion of ischemia-related and adenosine-responsive VTs would potentially narrow applicability.
The trial used drug doses that deviated from those recommended by current guidelines.1, 4, 5 The amiodarone dose in the study (5 mg/kg over 20 minutes) is similar to the ERC recommendation5 but higher than the AHA’s 150 mg (2 mg/kg for a 75-kg patient) over 10 minutes, but a repeat dose would be comparable over a 20-minute period.1, 4 This may have resulted in more adverse events due to a larger dose of amiodarone being administered rapidly than if practicing according to AHA guidelines assuming that the patient would respond to the first 150-mg bolus.1, 4 On the other hand, the procainamide dose (10 mg/kg over 20 minutes) is about in the middle of the 20 to 50 mg/ min range recommended by AHA, although practicing according to AHA guidelines would have continued the infusion past 20 minutes to a maximum dose of 17 mg/kg either until rhythm conversion or until an adverse effect (hypotension, QRS prolongation > 50%) occurred.1, 4 This procainamide dosing in the trial may have resulted in both lower major adverse cardiac event rate and lower efficacy as compared to AHA dosing.
Finally, the study focused on major cardiac adverse events (defined as clinical hypoperfusion, hypotension, signs of heart failure, increase in tachycardia, or development of polymorphic VT) as a primary outcome, which we have reported as NNH. The benefit endpoint (conversion to the baseline rhythm) was only a secondary outcome, and the study was not powered to measure this outcome. This leaves the authors only able to conclude that procainamide resulted in fewer adverse events, while evidence of superior cardioversion efficacy is hypothesis-generating only.
In summary, the evidence from this RCT is weak. It may also be the best available data well into the future, as evidenced by the researchers’ laudable, ultimately Herculean, 6-year effort to generate even these data. While the results nominally favor the use of procainamide for pharmacologic conversion of stable VT and align with a recent review of the existing literature,13 a true answer will remain elusive until higher quality RCTs are performed. Electrical cardioversion likely remains the most effective therapy for VT.1, 2 Whenever possible, the risks and benefits should be discussed with the patient and the choice of electrical versus pharmacologic cardioversion be made on a case-by-case basis. For this reason, we have assigned a color recommendation of “yellow” (unclear if benefits) to this summary.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Kyle Kelson, MD; Ian deSouza, MDSupervising Editor: Kabir Yadav, MD
Published/Updated
References:
Tranexamic Acid for the Treatment of Epistaxis
Benefits in NNT
5
NNT of 5 to prevent one episode of rebleeding
Harms in NNT
No difference in adverse events was reported
View As:
Source
Joseph J, Martinez-Devesa P, Bellorini J, Burton MJ. Tranexamic acid for patients with nasal haemorrhage (epistaxis). Cochrane Database Syst Rev 2018;12:CD004328.Study Popoulation: Six trials comprising 692 adult patients with epistaxis
Efficacy Endpoints
Rebleeding within 10 daysHarm Endpoints
Serious adverse events such as seizure and thromboembolic events; minor adverse events such as nausea, vomiting, or intoleranceNarrative
Epistaxis is a common reason for patients to present to the emergency department (ED), reflecting one of every 200 ED visits in the United States.1 While many cases of epistaxis are self-limiting, those requiring medical treatment can be associated with significant time and health care costs.2 Additionally, nasal packing and hemostatic matrices can be painful and require the patient to return for at least one follow-up visit. Therefore, identifying an effective and inexpensive treatment is of particular importance. Tranexamic acid is an antifibrinolytic agent that has been proposed as one potential modality for this.The Cochrane Review discussed here included randomized controlled trials comparing tranexamic acid (TXA) in any formulation (e.g., delivered orally, intravenously, or topically) with usual care versus usual care with placebo, usual care with any other hemostatic agent, or usual care alone.3 The primary outcome was the proportion of patients with rebleeding within 10 days and significant adverse events (i.e., seizures, thromboembolic events). Among the six trials (n = 692 patients), two studies used oral TXA,4, 5 while the remaining four used topical TXA.6, 7, 8, 9 For the primary outcome (n = 225 patients), TXA was associated with lower rates of rebleeding at 10 days (47% vs. 67%; relative risk [RR] = 0.71, 95% confidence interval = 0.56 to 0.90; absolute risk difference = 20%; number needed to treat = 5; moderate-quality evidence) compared to placebo. There were no significant differences between groups for adverse events, although only five of the trials reported adverse events.4, 5, 7, 8, 9 The included trials did not report outcomes requiring further intervention (e.g. repacking, surgery, embolization).
Another recent systematic review of topical TXA in epistaxis identified faster discharge rates, reduced rebleeding at 24 hours, and greater patient satisfaction with TXA, but no difference in rebleeding at 30 minutes.10 While the studies utilized different search strategies, both were informed by similar studies in their reviews. The current review further supports the potential value of this intervention.10
Caveats
Interpreting the results of the systematic review and meta-analysis discussed here warrants some caution. First, there was significant clinical heterogeneity in the study populations, with differences in the routes of administration (i.e., oral vs. topical), comparator groups (placebo vs. anterior nasal packing), and primary outcomes. Additionally, only three studies assessed the primary outcome of rebleeding at 10 days, while several other trials had different individual study outcomes (e.g., bleeding control within 30 minutes). Moreover, there was poor reporting of adverse events in the included studies. However, no significant adverse events were reported and most events were considered minor in nature (e.g., nausea, vomiting). Further, while rebleeding at 10 days is a clinically significant outcome, there were limited data on other ED-relevant outcomes (e.g., time to bleeding cessation, time to discharge, return to ED rates). Anterior epistaxis might also respond differently to treatment than posterior epistaxis. Only three of the included trials assessed the location of bleeding and only enrolled patients with anterior epistaxis. Other trials did not specify the location of bleeding. Another important limitation is that a large number of patients in some of the trials were on antiplatelet agents (i.e., aspirin, clopidogrel, or both); this could have affected the outcomes and contributed to the clinical heterogeneity in response to treatment. Future studies should identify what subgroup of patients (e.g., anterior vs. posterior epistaxis, antiplatelets use) are most likely to benefit from TXA, which delivery route is most effective, and how to better assess differences in adverse events.The existing evidence supports the efficacy of TXA to reduce the risk of rebleeding at 10 days among adult patients. Despite inconsistent reporting of adverse events, the occurrence of such events appears to be unlikely, particularly with topical use. Therefore, we have assigned a color recommendation of green (benefit > harm) to the use of TXA for epistaxis.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Michael Gottlieb, MD; Alex Koyfman, MD; Brit Long, MDPublished/Updated
May 3, 2019References:
Antibiotics for Acute Sinusitis in Adults
Benefits in NNT
17
1 in 17 were helped (cure at 7-14 days)
Harms in NNT
8
1 in 8 were harmed (adverse medication effects)
View As:
Source
Lemiengre MB, van Driel ML, Merenstein D, Liira H, Mäkelä M, De Sutter AIM. Antibiotics for acute rhinosinusitis in adults. Cochrane Database of Systematic Reviews 2018, Issue 9. Art. No.: CD006089. DOI: 10.1002/14651858.CD006089.Study Population: 3057 participants suspected of acute rhinosinusitis (symptoms for 30 days or less) from 15 trials in ambulatory care settings
Efficacy Endpoints
Clinical cure (resolution or improvement of major symptoms)Harm Endpoints
Adverse effects from antibiotic useNarrative
Acute sinusitis is a common condition encountered by clinicians in the ambulatory and emergency department setting. It is characterized by inflammation of nasal passages and sinuses, resulting in purulent nasal discharge, sinus tenderness, and facial pain. A majority of these cases are caused by a viral or self-limiting bacterial infection neither of which require antibiotics for treatment.1 Despite longstanding guidelines that recommend limiting the use of antibiotics to a small subset of patients, a majority of patients continue to be prescribed antibiotics.2This Cochrane Review discussed here3 examined data on benefits and harms associated with the use of antibiotics in adults diagnosed with acute rhinosinusitis (symptoms for 30 days or less). The rate of cure without any antibiotic use was 46% after one week and 64% after two weeks. The definition of cure varied depending on study but in most cases was defined as resolution or improvement of major symptoms.
The Cochrane review categorized the participants into three groups: those with clinically-diagnosed, radiographically diagnosed, and computerized tomography (CT)-diagnosed rhinosinusitis. The use of antibiotics was associated with a significant increase in the cure rate of clinically diagnosed rhinosinusitis (odds ratio [OR]: 1.25, 95%CI, 1.02 to 1.54; absolute risk difference [ARD]: 5%; Number-needed-to-treat [NNT]: 19; high quality evidence), as well as radiographically diagnosed (OR: 1.57, 95% CI, 1.03 to 2.39; ARD: 10; NNT:10; moderate quality evidence) and CT diagnosed rhinosinusitis (OR: 4.89, 95%CI, 1.75 to 13.72; ARD:25%, NNT:4; moderate quality evidence; one trial).3 Overall, antibiotics increased the rate of cure by 6% (absolute risk increase) corresponding to a number-needed-to-treat of 17 (OR: 1.38, 95%CI, 1.15 to 1.65).3
Adverse events were significantly increased with the use of antibiotics (OR:2.21, 95%CI, 1.60 to 2.77; ARD: 12.5%; number-needed-to-harm [NNH]:8). The type of adverse events varied with the most common being gastrointestinal effects such as diarrhea.3
Secondary outcomes such as resolution of purulent secretion, resolution of pain, illness duration, and restriction of daily activities could not be quantitatively assessed due to data heterogeneity.
Caveats
The findings here might not be generalizable to all patients with sinusitis. The meta-analysis excluded or did not consider patients with severe symptoms, pediatric patients, immunocompromised patients, or those with chronic symptoms.There was a higher cure rate in patients diagnosed by CT scan. But the usefulness and generalizability of this particular finding are limited by the fact that the data were derived from only a single trial. Moreover, imaging is not routinely used in patients with acute rhinosinusitis, and patients who require imaging (e.g. those with chronic sinusitis) may respond differently to antibiotics than the patients more commonly seen in emergency departments or outpatient offices. For most patients, the cost and the risk of radiation exposure outweigh the benefit of routine imaging for diagnosis, even if in fact this is useful in identifying individuals who are more likely to be helped by antibiotics.
Some of the more serious adverse effects of antibiotics that are uncommon or more difficult to quantify (e.g. allergic reactions, C. difficile infection, and the development of antibiotic resistance) were not reported in this analysis.
American Academy of Otolaryngology-Head and Neck Surgery4 and Infectious Disease Society of America5 both recommend observation and symptomatic management similar to that of acute viral rhinosinusitis for acute uncomplicated sinusitis. These guidelines recommend antibiotics in patients who fail symptomatic management after approximately 7 days.4, 5
We thus assign a color recommendation of Red (risk of harm exceeds potential benefit) for this treatment because of the relatively high rate of reported adverse effects and the high likelihood of resolution of symptoms with supportive care and symptomatic management. Our recommendation is in line existing guidelines for selective antibiotic use in this condition.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
See theNNT.com's previous reviews of this topic:
Antibiotics for Clinically Diagnosed Acute Sinusitis in Adults, January 8, 2013
Antibiotics for Radiologically-Diagnosed Acute Maxillary Sinusitis, January 8, 2013
Antibiotics for Acute Maxillary Sinusitis, September 23, 2010
Author
Nathan Franck, MD; Shahriar Zehtabchi, MDPublished/Updated
References:
Intensive Glucose Control for Critically Ill Patients
Benefits in NNT
No significant benefits
Harms in NNT
12
12 for severe hypoglycemia
View As:
Source
Fu Y, Sun Y, Zhang J, et al. Intensive glucose control for critically ill patients: an updated meta-analysis. Endocr Connect 2018;7:1288–98.Study Population: 17,582 adults hospitalized in critical care settings and enrolled in 27 trials
Efficacy Endpoints
3- to 6-month mortality, short-term mortality, sepsis, new dialysisHarm Endpoints
Severe hypoglycemiaNarrative
In the past decade, emergency department (ED) to intensive care unit (ICU) admissions increased by 79% to 2.2 million admissions annually, reflecting the increasing role of emergency medicine physicians in providing care for critically ill patients.1 Optimal glucose control in critical care patients has been a topic of contention for decades. In 2001 a single-center trial of mechanically ventilated surgical patients found intensive glucose control (maintaining glucose at 80–110 mg/dL) reduced mortality compared to conventional control (180–200 mg/dL only if glucose exceeded 215).2 Subsequent studies provided conflicting data, and in 2009, the multicenter NICE-SUGAR trial, the largest trial yet, demonstrated increased mortality with intensive glucose control.3 Current American Diabetes Association (ADA) guidelines, reflecting concern about harms associated with intensive glucose control, recommend conventional glucose control with a target glucose range of 140 to 180 mg/dL for critically ill patients who experience persistent hyperglycemia.4The meta-analysis summarized here provides an updated review of intensive glucose control effects on critical care patients.5 A total of 27 randomized trials enrolling 17,582 patients compared intensive with conventional glucose control in adult medical, surgical, and mixed critical care settings. Most had similar glucose targets. The primary outcomes were 3- to 6-month and short-term mortality (mainly within 28 days). Secondary outcomes were severe hypoglycemia (defined as serum glucose < 40 mg/dL: associated with increased mortality in multiple studies),6, 7, 8 sepsis, and need for dialysis.
There was no significant difference found in any primary outcome, and among secondary outcomes, only severe hypoglycemia in the intensive group was more common (relative risk = 4.9, 95% CI = 3.2– 7.5, NNH = 12). Notably, there was no significant difference found in any outcome between patients in medical, surgical, or mixed ICUs.
Caveats
This meta-analysis is limited in several ways. There was variation, in glucose targets, type of insulin, dose and mode of administration, duration of follow-up, and concomitant therapy. Additionally, not all trials reported on all outcomes of interest, and patient-level data are not available, limiting secondary research.The quality of evidence included in this meta-analysis is high. For most outcomes, despite the clinical heterogeneity noted above, there was little statistical heterogeneity. The only outcome with significant heterogeneity was severe hypoglycemia (I2 = 76.1%, p < 0.001), suggesting that clinical variation between studies affected this outcome.
Despite these flaws three additional, slightly less recent reviews have pooled these data as well with similar results despite differing numbers of trials, subjects, and point estimates. This consistency across author groups and approaches is reassuring.9, 10, 11
This meta-analysis also fails to address some ongoing research that has identified subgroups of patients who may stand to benefit from intensive glucose control. For example, two recent studies from the surgical ICU setting have found that among nondiabetic patients who had undergone major cardiothoracic surgery, intensive glucose control reduced morbidity.12, 13 No similar benefit was found for patients with a prior diagnosis of diabetes. Despite these interesting findings and ongoing research, conventional glucose control currently remains the standard of care in hospitalized patients.14
In summary, there was no benefit found with intensive glucose control in critical care patients but there was increased incidence of severe hypoglycemia. With no benefits and increased harms, the most appropriate color rating for intensive glucose control is black (harms > benefits). Current ADA guidelines, citing the findings of prior meta-analyses, recommend conventional glucose control with targeted blood glucose of 140 to 180 mg/dL in critically ill patients who experience persistent hyperglycemia.4
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
John Conway; Benjamin Friedman, MDSupervising Editor: Shahriar Zehtabchi, MD
Published/Updated
References:
Ticagrelor Compared to Clopidogrel in Acute Coronary Syndrome and Stable Coronary Artery Disease
Benefits in NNT
No patients were helped
Harms in NNT
166
1 in 166 patients had a major bleeding event
13
1 in 13 patients developed dyspnea
View As:
Source
Guan W, Lu H, Yang K. Choosing between ticagrelor and clopidogrel following percutaneous coronary intervention: A systematic review and Meta-Analysis (2007-2017). Medicine. 2018;97(43):e12978.Study Population: Patients with acute coronary syndrome (ACS) who underwent percutaneous coronary intervention (PCI)
Efficacy Endpoints
Death, heart attack, strokeHarm Endpoints
Major bleeding and dyspneaNarrative
Dual antiplatelet therapy with compared to aspirin alone after percutaneous coronary intervention modestly reduces nonfatal events like heart attack and stroke.1, 2 This has led some to believe better antiplatelet agents could improve outcomes further, and ticagrelor has been proposed as such an agent.The systematic review summarized here3 included 11 trials and 5 observational studies that in aggregate enrolled 25,805 subjects with acute coronary syndrome undergoing percutaneous coronary intervention as well as those with stable CAD. The patients included were predominantly male, with mean age 54-72 years old. The trials enrolled patients from United States, United Kingdom, Japan, Korea, Spain, Italy, France, Taiwan, and China.
Ticagrelor, as compared to clopidogrel, did not reduce heart attacks, strokes, deaths, or stent thrombosis, but did increase major bleeding (absolute risk difference [ARD]: 0.6%; odds ratio [OR]: 1.52; 95%CI, 1.01 - 2.29; Number-needed-to-harm [NNH]: 166). Major bleeding was defined as significant drop in hemoglobin (>3 g/dl) or requiring blood transfusion, or intra-ocular bleeding resulting in visual loss or complete blindness.3
More patients in the ticagrelor group experienced dyspnea (ARD: 7.6%, OR: 2.64, 95%CI, 1.87 - 3.72; NNH: 13), which could be one reason why there was a significantly higher rate of drug discontinuation in ticagrelor group (ARD: 1.7%, OR:5.67, 95%CI, 1.26 - 25.54; NNH: 59).3
Caveats
The evidence quality was variable, but most notably 5 observational studies were included. This is a methodologically important error, in our opinion. It is compounded by pooling them with trial data. This calls into question all results, particularly those which tended to hover near statistical significance. The observational data may have diluted or enhanced any effect. Some trials also included patients who suffer from stable CAD. However, a subgroup analysis that did not include these patients found similar results.While based on the guidelines, patients undergoing PCI should be on dual antiplatelet therapy (asprin plus clopidogrel or ticagrelor), not all trials reported the exact number of patients on dual antiplatelets. However, the ones who reported the numbers, had nearly 100% of the patients on dual antiplatelet regimen.
Moreover, some individual trials in the meta-analysis report benefits for heart attack and mortality. The largest (PLATO, funded by ticagrelor’s manufacturer) included over 18000 subjects, finding a statistically significant 1.1% benefit for heart attack and 1.4% for mortality. Unlike most in the meta-analysis the trial followed the subjects for 12 months.4 This raises another limitation, follow up periods. Some reported one-month or in-hospital outcomes. Perhaps, therefore, short follow-up underestimated efficacy. This notion is challenged, however, by a nearly identical study of 800 subjects with 12 months follow-up, also funded by the maker of ticagrelor, which attempted to reproduce PLATO in an Asian setting, failed to find any benefit, and bordered on showing overall harm.5
Notably, ticagrelor (brand name Brilinta) is much more expensive than clopidogrel (Plavix). As of March 2019, the price for a one month supply of each was approximately $400 and $11, respectively.6
The 2016 ACC/AHA Guideline “Focused Update on Duration of Dual Antiplatelet Therapy in Patients With Coronary Artery Disease,” makes a class IIa recommendation that ticagrelor is preferential to clopidogrel for both non-ST-elevation acute coronary syndrome and ST-elevation myocardial infarction and patients who undergo percutaneous coronary intervention. We believe that the existing evidence does not support this recommendation, as there was no statistically significant benefit in stent re-thrombosis or all-cause mortality, while there was evidence of increased risk of harm compared to clopidogrel.7
In summary, the existing evidence is poor, includes observational data pooled with trial data, and finds only harm, no benefit, with ticagrelor. We have assigned a color recommendation of Red (No Benefits) because of the uncertainty about benefits and evidence of potential harms. We hope to see a more rigorous review as well as further relevant trials in the near future.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Donald Doukas, MD; Hannah Schimmer, NP; Shahriar Zehtabchi, MDPublished/Updated
April 15, 2019References:
Denosumab for Reducing Risk of Fractures in Postmenopausal Women
Benefits in NNT
21
1 in 21 did not have a new vertebral fracture
62
1 in 62 did not have a new clinical vertebral fracture
230
1 in 230 did not have a new hip fracture
Harms in NNT
167
1 in 167 developed an infection
View As:
Source
Zhou Z, Chen C, Zhang J, Ji X, Liu L, Zhang G, Cao X, Wang P. Safety of denosumab in postmenopausal women with osteoporosis or low bone mineral density: a meta-analysis. Int J Clin Exp Pathol. 2014 Apr 15;7(5):2113-22. eCollection 2014.Cummings SR, San Martin J, McClung MR, Siris ES, Eastell R, Reid IR, et al.; FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009 Aug 20;361:756-65.
Study Population: Postmenopausal women with documented osteoporosis
Efficacy Endpoints
Prevention of new vertebral and non-vertebral fracturesHarm Endpoints
Infection, neoplasm, deathNarrative
As bone density decreases people are at an increased risk for fractures.1 Denosumab is a fully human monoclonal antibody that binds the receptor activator of nuclear factor-κB ligand (RANKL), which prevents its interaction with the osteoclast and osteoclast precursor surface receptor, RANK. This inhibits osteoclast-mediated bone resorption by blocking osteoclast function, formation, and survival.2, 3 Prior studies have demonstrated an increase in bone mineral density with the administration of denosumab in post-menopausal women.4, 5 This review assesses whether the increase in bone mineral density translates into a reduction in the risk of osteoporosis-related fractures.The FREEDOM trial2 is the largest randomized control trial to compare denosumab vs placebo in the prevention of fractures in postmenopausal women with osteoporosis. Women between the ages of 60 and 90 years with a bone mineral density T score of less than -2.5 (consistent with the typical definition of osteoporosis) at the lumbar spine or total hip were included in the trial. Patients were randomly assigned to receive subcutaneous injections of either 60 mg of denosumab or placebo every 6 months for 36 months. In this trial, the primary endpoint was new vertebral fractures based on semi-quantitative grading scales of lateral spine radiographs.2 The treatment with denosumab was associated with significantly lower risk of new vertebral fractures (Relative risk [RR]: 0.32, 95% confidence interval [CI], 0.26 - 0.41]; Absolute risk difference [ARD]: 4.8%; NNT 21). Secondary outcomes included non-vertebral fractures (NNT 71), hip fractures (NNT 230), new clinical vertebral fractures (NNT 62), and multiple (≥2) new vertebral fractures (NNT 103). The study found no significant difference in the incidence of infection, death, or neoplasm.2
A meta-analysis3 published in 2014 examined the safety of denosumab in 15,263 postmenopausal women from 13 trials with documented osteoporosis who were followed between 9 months to 3 years after initiation of therapy. This meta-analysis found a non-significant reduction in the risk of non-vertebral fractures with the administration of denosumab (RR: 0.86, 95%CI, 0.74 - 1.00; ARD: 0.83%; NNT 121). The meta-analysis found the difference in incidence of death or neoplasm to be non-significant. However, a non-statistically significant difference in rates of infection (RR: 1.23, 95%CI, 1.00 - 1.52; ARD: 0.60%; NNH 167) was seen.2 We used this information to calculate the number-needed-to-harm (NNH) because the sample size for the meta-analysis was larger than the FREEDOM trial2 and also the objective of the meta-analysis specifically was to assess the safety of the treatment.3 Another systematic review6 published in 2014 compared the efficacy of various pharmacologic treatments in reducing the risk of fractures. This analysis also confirmed the efficacy of denosumab in reducing the risk of fractures in postmenopausal women. For different pharmacologic treatments including various bisphosphonates, bisphosphate derivatives, teriparatide, raloxifene, or denosumab; NNT for vertebral fractures was in the range of 60-89 and NNT for non-vertebral fracture was 50-60.6 The review also found denosumab to have an NNH of 118 for infection.6
Caveats
While the meta-analysis by Zhou et al3 included 11 randomized controlled trials, the FREEDOM trial2 was by far the largest study with their sample size accounting for about 60% of the total number of participants in all studies.While the existing evidence supports the safety of denosumab, the follow-up period for the trials (ranging from 9 months to 3 years) may have been too short for assessing the harm endpoints of neoplasm, death, or infection. Longer-term follow-ups are needed to understand the long-term safety profile of this treatment. One longer-term follow-up was for the patients in the FREEDOM trial who were followed for an additional 7 years, and in this follow up study, the rates of serious adverse events for the participants treated with denosumab remained low (11·5 and 14·4 per 100 participant-years).7 Denosumab is marketed under brand names Prolia and Xgeva. As of October 2018, the price of one 60 mg syringe or vial, which has to be administered every 6 months, is approximately $1200-1400.8
The manufacturer’s website lists some examples of possible infections associated with it as infections of “skin, lower stomach, bladder, ear, or the inner layer of the heart (endocarditis)”.9
In conclusion, denosumab appears to be effective in reducing the risk of vertebral and non-vertebral fractures in premenopausal women with osteoporosis. Treatment with denosumab does not increase the risk of cancer or death but might increase the risk for infection. However, given the non-statistically significant impact on non-vertebral fractures and the uncertainty of longer-term harms due to relatively short follow up periods, we assign a color recommendation of Yellow (unclear benefit; more studies required) to this treatment.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Peter Song, MD; Shahriar Zehtabchi, MDPublished/Updated
References:
Omega-3 Fatty Acids and Cardiovascular Disease Prevention
Benefits in NNT
No deaths from any cause, deaths from cardiovascular disease, cardiovascular events, arrhythmias or strokes were prevented
Harms in NNT
14
1 in 14 experienced nausea
View As:
Source
Abdelhamid AS, Brown TJ, Brainard JS, et al. Omega-3 fatty acids for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst Rev 2018;7:CD003177.Study Population: Adults (age ≥18 years) with varying levels of cardiovascular risk
Efficacy Endpoints
All-cause mortality, cardiovascular mortality, cardiovascular events, arrhythmia, strokeHarm Endpoints
Nausea, abdominal pain or discomfort, diarrhea, reflux, any gastrointestinal side-effect, headache or worsening migraine, joint and muscle pain, skin problems, gastrointestinal hospitalization, bleeding, pulmonary embolus or deep venous thrombosisNarrative
Non-communicable diseases have overtaken communicable diseases as the world's major disease burden, with circulatory and cardiovascular diseases (CVD) remaining the leading cause of death globally.1 Researchers have long investigated the health effects of omega-3 (Ω-3) polyunsaturated fatty acids (PUFA), including eicosapentaenoic acid (EPA; C20:5) and docosahexaenoic acid (DHA; C22:6), on CVD prevention. Dietary fatty acids may be oxidized for energy, stored in adipose tissue, or further metabolized to various long-chain PUFAs. Membrane derived PUFA’s serve as substrates for formation of eicosanoid effectors (Ω-3 and Ω-6).2 The effectors derived from Ω-3 PUFAs are less inflammatory and platelet aggregating than their Ω-6-derived counterparts.2The primary source of nonprescription Ω-3 PUFA supplements is fish oil.3 In 2012, 7.8% of U.S. adults (18.8 million) reported consuming a fish oil dietary supplement within the prior 30 days.4 An American Heart Association (AHA) report suggested that Ω-3 PUFA supplements may reduce coronary heart disease (CHD) death, possibly through a reduction in ischemia-induced sudden cardiac death (SCD), among patients with prior CHD, but does not reduce the incidence of recurrent nonfatal MI. As benefits likely outweigh the risks, the AHA report offered a Class IIa recommendation (Benefits >> Risk. Additional studies with focused objectives needed. It is reasonable to administer treatment) for the use of Ω-3 PUFA supplements for the secondary prevention of CHD death.3
The meta-analysis discussed here analyzes the effectiveness of dietary Ω-3 PUFA supplementation with EPA (fish-derived; C20:5), DHA (fish-derived; C22:6) and alpha-linolenic acid (ALA; plant-derived; C18:3) to improve all-cause mortality, cardiovascular deaths, cardiovascular events, arrhythmias, and stroke. The meta-analysis included 79 randomized controlled trials (RCT) enrolling 112,059 participants published before April 2017.5 Twenty-seven on-going trials were excluded. Thirty-three trials included subjects with prior cardiovascular risk (secondary prevention), and 46 trials enrolled patients with no prior cardiovascular risk (primary prevention). The follow-up period varied from 12 to 72 months. Ω-3 PUFA supplementation was performed via capsule or medicinal oils, Ω-3 PUFA-enriched foods, or via dietary advice to increase Ω-3 PUFA intake. Due to small number of trials assessing long-chain Ω-3 (LCn3) fatty acids, EPA and DHA were analyzed in aggregate.5
Benefits:
Long-Chain Polyunsaturated Fatty Acids
No significant benefits were noted with LCn3 PUFA (EPA, DHA) supplementation for preventing all-cause mortality (39 RCTs; 92,653 participants), cardiovascular mortality (25 RCTs; 67,722 participants), cardiovascular events (38 RCTs; 90,378 participants), arrhythmia (28 RCTs; 53,796 participants), or stroke (28 RCTs; 89,358 participants).
Alpha-Linolenic Acid (ALA)
No significant benefits were noted with ALA supplementation for preventing all-cause mortality (5 RCTs; 19,327 participants; 459 deaths), cardiovascular mortality (4RCTs; 18,619 participants), cardiovascular events (5 RCTs; 19,327 participants; 884 CVD events), arrhythmia (1 RCT; 4,837 participants), or stroke (5 RCTs; 19,327 participants).5
Harms:
The meta-analysis assessed both non-serious side-effects (nausea, abdominal pain or discomfort, diarrhea, reflux, any gastrointestinal side-effect, headache or worsening migraine, joint lumbar and muscle pain, or skin problems) and serious side-effects (gastrointestinal hospitalization, bleeding, pulmonary embolus (PE) or deep venous thrombosis (DVT).
Long-Chain Polyunsaturated Fatty Acids
There was no suggestion that LCn3 significantly increased non-serious side effects (aggregate) with high heterogeneity.5 Nausea was increased in the LCn3 PUFA group (RR 1.76, 95% CI 1.25 to 2.48; I2 =0%; 5 RCTs; 1,234 participants). All other non-serious side effects were not statistically different.5
Serious adverse events were not increased with LCn3 intake.
Alpha-Linolenic Acid
ALA supplementation did not increase side-effect mediated study withdrawal.5 Insufficient data was available to assess other non-serious adverse effects.
Data was insufficient for assessing the risk of PE or DVT (1 RCT; 708 participants; 1 event). No data was available for any other serious adverse events.
Caveats
Ω-3 PUFA enriched foods (e.g. dietary fish) may have different health effects than capsule or medicinal oil Ω-3 PUFA, as it may replace less healthy foods (leading to reduced saturated fat intake) and provides other additional nutrients (e.g. protein, selenium, iodine, calcium, magnesium). The sub-group analysis performed in the meta-analysis was underpowered to detect a statistically significant difference between the dietary advice subgroups (enriched food vs. capsule or medicinal oil). Daily Ω-3 PUFA intake in such studies is not quantifiable as dietary practices vary across patients. Thus, it remains unclear whether studies using this design skewed the potential effects of studies utilizing a more traditional design. Furthermore, the assessment of EPA and DHA in aggregate potentially masked the benefit of any singular agent.The follow-up duration of at least 12 months may not have been long enough to detect any significant impact on mortality and cardiovascular outcomes.
Evidence from funnel-plots reported in the meta-analysis suggest some small study-bias, suggesting some smaller studies showing increased risk of CVD outcomes with Ω-3 PUFA use may be missing. Additionally, the data is limited by significant heterogeneity. This likely reflects the variable means by which data points were collected across trials.
Lastly, this analysis5 did not collate data on cancers and neurological problems associated with polychlorinated biphenyls (PCBs) or mercury in fish oils.
Conclusion: In summary, high-quality evidence suggests that long-chain Ω-3 PUFA do not prevent mortality (all-cause or cardiovascular) or cardiovascular disease events when used as primary or secondary prevention. Because of lack of any benefit but relative safety, we have assigned a color recommendation of RED (no benefit) to this treatment.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Anna Tomdio, MD; Michael Ritchie, MD; Andrew C Miller, MDPublished/Updated
References:
Flexible Sigmoidoscopy or Fecal Occult Blood Testing for Colorectal Cancer Screening in Asymptomatic Individuals
Benefits in NNT
450
Flexible sigmoidoscopy: 1 in 450 did not die from colorectal cancer
900
Fecal occult blood testing: 1 in 900 did not die from colorectal cancer
Harms in NNT
1250
Flexible sigmoidoscopy: 1 in 1250 experienced an adverse outcome
3300
Fecal occult blood testing: 1 in 3300 experienced an adverse outcome
View As:
Source
Holme Ø, Bretthauer M, Fretheim A, Odgaard-jensen J, Hoff G. Flexible sigmoidoscopy versus faecal occult blood testing for colorectal cancer screening in asymptomatic individuals. Cochrane Database Syst Rev. 2013;(9):CD009259.Study Population: Asymptomatic adults (45-80 years old)
Efficacy Endpoints
Death from colorectal cancer, death from any causeHarm Endpoints
Bleeding, perforation, and death resulting from follow-up colonoscopy or surgeryNarrative
Colorectal cancer (CRC) continues to be the third leading cause of cancer death in the United States.1, 2 The aim of screening is to reduce mortality through early detection.2, 3There are several methods available for colorectal cancer screening, such as stool-based test (guaiac fecal occult blood test and fecal immunochemical test), endoscopic methods (sigmoidoscopy and colonoscopy), and imaging methods with computed tomographic colonography among other new techniques.4 Fecal occult blood testing (FOBT) and flexible sigmoidoscopy has not been directly compared to determine the superior screening modality, if present.1
The Cochrane meta-analysis cited here assessed the effectiveness of FOBT and flexible sigmoidoscopy as a colorectal cancer (CRC) screening modality in an asymptomatic population.1 The primary outcome measured was colorectal cancer mortality.1 The meta-analysis included 9 randomized controlled trials (RCTs; 338,467 participants in the screening group and 405,919 in the control group). Flexible sigmoidoscopy as a screening modality was compared to no screening in 5 RCTs (165,733 of 414,744 participants); and guaiac-based FOBT was compared to no screening in 4 RCTs (172,734 of 329,642 participants).1 Colorectal cancer mortality was lower with flexible sigmoidoscopy (Relative risk [RR]:0.72, 95% Cl 0.65 to 0.79; Absolute Risk Difference [ARD]: 0.22%; NNT:450) and FOBT (RR 0.86, 95% Cl 0.80 to 0.92; ARD: 0.11%; NNT:900) when compared with no screening.1 In other words, one would need to screen 500 patients with flexible sigmoidoscopy to prevent 1 death resulting from colorectal cancer; whereas, one would need to screen 900 patients with FOBT to prevent 1 colorectal cancer death.1 The latter can be explained with increased follow-up colonoscopies for positive FOBT results.1 Despite this fact, neither of these modalities reduce all-cause mortality (7 trials).1
Major complications (e.g. bleeding, perforation, or death) within 30 days of screening, follow-up colonoscopy or surgery occurred in 0.03% and 0.08% of participants in the FOBT and flexible sigmoidoscopy trials, respectively.1
Caveats
Despite evidence of flexible sigmoidoscopy and FOBT reducing colorectal cancer mortality, there are caveats to consider.1 Unfortunately, the meta-analysis does not provide a clear answer regarding a superior screening modality.1 In the absence of direct evidence, it cannot be concluded whether the net benefit or harm of one modality is greater than the other.1 The decision to choose one test over another should be a shared decision made by the patient and the physician. In a patient with no family history of colon cancer, FOBT as the less invasive approach could be the first logical choice. The U.S. Preventive Services Task Force guidelines recommend colon cancer screening for adults between the ages of 50-75.5In conclusion, this meta-analysis provides high quality evidence that flexible sigmoidoscopy and fecal occult blood testing both reduce the risk of death from colorectal cancer.1 Therefore, we assign a color recommendation of Green (Benefits>Harm) to both screening tests.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Karissa A. Lambert MD; Ahmed Hamed MD; Amira Hamed MDPublished/Updated
March 29, 2019References:
Low-dose CT Scan for Lung Cancer Screening
Benefits in NNT
219
1 in 219 may have been helped (one death from any cause was prevented) compared to screening with chest x-ray
Harms in NNT
19
1 in 19 were harmed (incidental finding on CT scan)
78
1 in 78 were harmed (false positive result on CT scan)
View As:
Source
Usman Ali M, Miller J, Peirson L, et al. Screening for lung cancer: A systematic review and meta-analysis. Prev Med. 2016 Aug;89:301-314. PMID: 27130532.Humphrey L, Deffebach M, Pappas M, et al. Screening for Lung Cancer: Systematic Review to Update the U.S. Preventive Services Task Force Recommendation. Rockville (MD): Agency for Healthcare Research and Quality (US); 2013 Jul. PMID: 24027793.
Study Population: Individuals at high risk for lung cancer
Efficacy Endpoints
Death from any causeHarm Endpoints
False positive results, incidental findings, complications of invasive procedures after a positive screening resultNarrative
Any means of reducing deaths due to lung cancer, the most frequently diagnosed cancer and leading cause of cancer death in the western world,1 would have major public health implications. We summarize two reviews of low dose computed tomography (LDCT) screening for lung cancer.2, 3Overall no mortality benefit was found in either review when comparing LDCT to usual care (no screening measures). Indeed two trials suggest LDCT worsens mortality.4, 5 However there is nearly a mortality benefit (RR 0.94; .95CI 0.88-0.998) when pooling these trials together with an additional large trial, NLST, that compares LDCT to chest x-ray for screening.6
Notably, NLST, or the National Lung Screening Trial, published in 2011,6 randomized >53000 high-risk people to screening with low dose chest CT (LDCT) or annual chest x-ray. As the only lung screening trial ever to demonstrate a benefit the study has been highly cited and led to some guidelines and recommendations to use LDCT screening.
Unlike screening for most other cancers in which screening is done on a general population lung cancer screening trials target smokers at high risk since lung cancers overwhelmingly occur in previous or current smokers. NLST subjects were 55-74 with an average 36 pack-year smoking history.
In the 7 year follow-up report for NLST mortality was 0.46% lower with LDCT (NNS: 217),7 an encouraging and oft-cited finding.
Caveats
There are reasons to be wary of the NLST findings. Most obviously it is a single trial while meta-analyses, like the reviews for this summary, found no mortality benefit even including NLST.* This is because earlier trials find no benefit and in two studies mortality was increased, counterbalancing NLST.Why the difference? As is often the case with multiple trials from multiple settings the answer is fuzzy. It may be the comparator: NLST compared LDCT to chest x-ray, which seems to be worse than nothing. Prior reviews including the two cited here find x-rays, when compared to nothing, cause false positives without saving lives and may lead to harms. LDCT could shine in comparison, even with a neutral or negative impact.
Or perhaps NLST was an anomaly. Almost 30% of the mortality advantage was made up of deaths unrelated to lung cancer. No explanation has been proffered for this, and LDCT did not seem to reduce deaths from other cancers or cardiovascular problems. Assuming this part of the difference is due to chance, removing it neutralizes the advantage (p=0.11).8
Conversely, the Canadian task force review, like the USPSTF, makes a “weak” recommendation for offering LDCT.4 These recommendations may or may not be premature. The Cochrane review points to short follow-up (7 years) and poorly understood harms. We are concerned there is a long history of irrational exuberance—often followed by reversal and regret—based on single studies not yet retested or reproduced.9
Challenges also exist for implementation of LDCT screening. For one, the cost will add billions to national health care expenditures annually,6 and while shared decision making has been mandated for LDCT this is largely being circumvented in practice.10
Although LDCT is the only screening strategy showing mortality benefit in a study it is associated with over-diagnosis and false positives, and the attendant physical and psychological consequences of both. Given the checkered history of LDCT, particularly when compared to usual care, the NLST results must be reproduced by other groups and in other settings before it can be considered a proven intervention.
Based on the (admittedly hopeful) possibility of benefit we have given this intervention a Yellow color designation, indicating more research is required.
*Editorial note: it is our policy to report, whenever possible, all-cause mortality in favor of disease-specific mortality, as the former is far more patient-centered. For those interested, in this case LDCT had a similar (no benefit) impact on ‘lung cancer mortality’.
Author
Saed Awadallah, MD; Michael Ritchie, MDSupervising Editor: Shahriar Zehtabchi, MD
See theNNT.com's previous reviews of this topic:
CT Scanning for Lung Cancer Screening in High-Risk Smokers, August 7, 2011
Published/Updated
References:
Corticosteroids for Treating Pneumonia
Benefits in NNT
17
NNT of 17 for preventing death in patients with severe pneumonia (pneumonia severity index score >4 or equivalent)
33
NNT of 33 for reducing risk of new respiratory failure
5
NNT of 5 for reducing risk of shock
Shorter length of hospital stay by average of 3 days
Shorter length of intensive care unit stay by average of 2 days
Harms in NNT
11
NNH of 11 for elevated blood sugar (hyperglycemia)
View As:
Source
Stern A, Skalsky K, Avni T, Carrara E, Leibovici L, Paul M. Corticosteroids for pneumonia. Cochrane Database Syst Rev 2017;(12):CD007720.Study Population: 2,264 adults and children admitted to the hospital with community-acquired pneumonia from 17 trials
Efficacy Endpoints
All-cause 30-day mortality, risk of new respiratory failure, risk of shock, length of hospital and intensive care unit stayHarm Endpoints
Hyperglycemia, secondary infections, neuropsychiatric complications, and gastrointestinal bleedingNarrative
Pneumonia remains a major cause of morbidity and mortality in the United States.1 There is both theoretical and laboratory evidence that corticosteroids may have beneficial effects in pneumonia through local pulmonary and systemic effects.2, 3The data for this evidence-based summary are derived from a Cochrane meta-analysis by Stern et al.,4 which included 17 trials with 2,264 adult and children admitted to the hospital with community-acquired pneumonia (CAP). We will also discuss another systematic review by Briel et al.5 that included 1,500 patients from six randomized controlled trials.
The Cochrane systematic review4 included adults and children hospitalized with CAP with or without health care-associated pneumonia. Patients were randomized to receive corticosteroids (any dose and any duration) plus usual care or placebo plus usual care. The most common dose given was the equivalent of 40 to 50 mg of prednisone daily for 5 to 10 days. This meta-analysis4 showed that corticosteroids decreased 30-day all-cause mortality in adults with severe pneumonia defined as pneumonia severity index score >4 or equivalent with a number needed to treat (NNT) of 17 (Table 1). In the entire cohort of patients (severe and nonsevere pneumonia), administration of corticosteroids was associated with a lower risk of new respiratory failure (defined as requiring noninvasive or invasive mechanical ventilation) and a reduced risk of shock with NNT of 33 and five, respectively (Table 1). Length of hospital and intensive care unit stays were also shortened for all adult patients (mean difference = –3 days; 95% confidence interval [CI] = –5 to –0.9; mean difference = –2 days; 95% CI = –2.96 to –0.81, respectively).4
The only adverse event associated with the use of corticosteroids was hyperglycemia with number needed to harm (NNH) of 11 (Table 1).4 However, hyperglycemia was not clearly or consistently defined. The risks of secondary infections, neuropsychiatric complications, gastrointestinal bleeding, and other adverse events were not significantly different between the groups.4
Briel et al.5 conducted a patient-level analysis (Cochrane was a trial-level analysis) based on data from six trials and found that corticosteroids had no clear mortality benefit neither in all cases of pneumonia (odds ratio [OR] = 0.75, 95% CI = 0.46 to 1.21) nor in severe pneumonia (OR = 0.70, 95% CI = 0.44 to 1.13). This meta-analysis confirmed shorter length of hospital stay for patients who received corticosteroids by >1 day but the rate of 30-day pneumonia-related rehospitalization was higher (5% vs. 3%; OR = 1.85, 95% CI = 1.03 to 3.32; NNH = 45). Pneumonia-related rehospitalization was defined as recurrent pneumonia, other infection, pleuritic pain, adverse cardiovascular event, or diarrhea. Corticosteroid treatment was associated with higher incidence of hyperglycemia.5
Caveats
The main limitation of the evidence for mortality benefit of corticosteroids in severe pneumonia according to the Cochrane meta-analysis4 is the possibility of bias, as the only positive effect on mortality was found in trials with unclear risk of bias.4 For this reason, the Cochrane authors downgraded the level of evidence from high to moderate.There is also a notable discrepancy between the two meta-analyses for this outcome. One reason for this discrepancy is that Briel et al.5 only included six trials in the meta-analysis while Cochrane included 17 trials.4 Briel et al. excluded several trials because patientlevel data were not available.5 These six trials were all included in the Cochrane analysis. Therefore, the analysis by Briel et al.5 was performed on only a subgroup of patients included in the Cochrane data set.4 Given the similarity of the effect estimate for mortality rate in severe pneumonia between both meta-analyses as well as a wider 95% CI in the analysis by Briel et al. compared to that of Cochrane (OR = 0.70, 95% CI = 0.44 to 1.13; vs. OR = 0.58, 95% CI = 0.40 to 0.84), it is likely that the exclusion of the trials without patient-level data reduced the statistical power in the analysis by Briel et al.4, 5
For the outcome of rehospitalization in the meta-analysis of Briel et al., 5% of patients were rehospitalized in the corticosteroid group as opposed to 2.7% in the placebo group (total N = 1,368, full distribution of patients not available due to lack of data).5 On the surface this outcome appears appropriately powered but this is a composite outcome. Patients were rehospitalized for “other infection, pleuritic pain, cardiovascular disease event, diarrhea.”5 Therefore, we feel that the data supporting this negative outcome should be interpreted with caution and it is not specifically powered to measure rehospitalizations directly due to steroid administration.
For the outcome of reducing risk of shock, while the Cochrane review found that there was significant benefit in the steroid arm,4 this effect may be overstated. The pooled risk of development of shock in the control group of the meta-analysis was approximately 24% among 500 patients.4 This rate is significantly higher than the reported rate of patients with CAP requiring vasopressors in the literature (approximately 5%).6 However, three of four of trials included in this outcome analysis were conducted on patients with severe pneumonia, which carry a higher baseline risk of development of shock. Therefore, we suggest that the NNT of 5 should be considered as only applying to patients with severe pneumonia and not necessarily to all patients.
The patient-level analysis approach employed by Briel et al.,5 which usually is used to reduce heterogeneity, offers little advantage over the Cochrane analysis as the heterogeneity of this meta-analysis was already low (I² = 12%, fixed-effect model).4 In our opinion, because of lower risk of type II error and low heterogeneity, the Cochrane review offers a more robust and accurate data set and thus more reliable findings for mortality benefit of corticosteroids in severe pneumonia.4
Children were also included in the Cochrane review4 but they were excluded from most outcome analyses because of paucity of data. Although the Cochrane review rated the quality of evidence for children as high, there were only four studies that enrolled children.4 Additionally, there were no reported deaths in these four trials. Therefore, the findings of this meta-analysis could not be generalized to the pediatric population.
Lastly, patients with pneumonia and a history of chronic obstructive pulmonary disease (COPD) may benefit more from corticosteroids. The Cochrane review did not perform a subgroup analysis to examine the effect of COPD on the results (three of 17 trials excluded patients with COPD). But they performed a meta-regression analysis that did not show an association between the percentage of COPD patients in the trials and corticosteroid mortality benefit.4
Clinical Implications: While the evidence for the mortality benefit of corticosteroids for pneumonia is of moderate quality, there are other patient-centered benefits including shortened hospital stay, prevention of shock, and lower risk of respiratory failure. The combination of improvement in patient-centered outcomes, lower medical costs and resource utilization, and relative safety is desirable not only for patients, but for institutions as well. Therefore, we assign a color recommendation of green (benefits > harms) to this treatment. In summary, the existing evidence supports the use of corticosteroids in patients hospitalized with pneumonia. This recommendation is based on a probable mortality benefit and lower risk of shock in patients with severe pneumonia as well as shorter length of hospital stay and respiratory failure in all patients. Low costs and low risk of adverse events augment these clinical benefits.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Peter Tepler, MD; Shahriar Zehtabchi, MDSupervising Editors: Gary Green, MD; James McCormack, MD
See TheNNT.com's previous review of this topic: Corticosteroids for Community-Acquired Pneumonia, June 1, 2016
Published/Updated
References:
Corticosteroids for Preventing Postherpetic Neuralgia After Herpes Zoster Infection
Benefits in NNT
None were helped
Harms in NNT
None were harmed
View As:
Source
Han Y, Zhang J, Chen N, He L, Zhou M, Zhu C. Corticosteroids for preventing postherpetic neuralgia. Cochrane Database Syst Rev 2013;(3):CD005582.Study Population: 787 patients with herpes zoster (shingles) infection who were randomized to treatment with corticosteroid or placebo in five trials
Efficacy Endpoints
Presence of postherpetic neuralgia 6 months after onset of initial acute herpetic rashHarm Endpoints
“Serious” (life-threatening) or “nonserious” adverse events during treatment or within 2 weeks after stopping treatmentNarrative
Postherpetic neuralgia (PHN) is a painful condition of persistent chronic pain following acute reactivation of varicella zoster virus. The review defines PHN as persisting or recurring pain at the site of shingles at least 1 month after the onset of the acute rash. The incidence of shingles increases with age, almost doubling in each decade after 50 years of age. Of these cases, roughly 20% go on to develop PHN, with age again being the strongest risk factor.1 The pain of PHN is frequently debilitating and can significantly affect quality of life. It is thought that the anti-inflammatory effects of corticosteroids might decrease nerve damage and prevent PHN.This Cochrane review is an update of a previous Cochrane review first published in 2008 and updated in 2010. This update concludes based on moderate quality evidence that steroids do not provide benefit in the prevention of PHN, whereas prior reviews indicated insufficient evidence to draw a conclusion. More up-to-date data analysis methods were used in this review to provide conclusions It included all randomized controlled trials in which corticosteroids were given by oral, intramuscular, or intravenous routes within 7 days after onset of rash and in which steroids were compared to either no treatment or to placebo. Five trials with a total of 787 patients were included. The meta-analysis provides moderate-quality evidence that corticosteroids are not effective in preventing PHN 6 months after onset of acute herpetic rash (relative risk = 0.95, 95% confidence interval = 0.45 to 1.99). The review found no statistically significant difference in the secondary outcome of pain severity at 3, 6, or 12 months.
Nonserious adverse events were recorded in all of the trials, and there were no statistically significant differences between steroid and placebo. Serious events, including pneumonia, myocardial infarction, cardiac insufficiency, and death, for example, were reported in three of the trials, but there were no statistically significant differences between steroid and placebo (6/376 [1.6%] and 3/379 [0.8%], respectively). Two of the trials made note of the absence of serious adverse events in the steroid groups.
The Cochrane authors suggest that future trials should include measurements of function and quality of life and, furthermore, that there should be longer term follow-up to determine an effect of steroids on the likelihood of transition from acute pain to PHN.
Caveats
Of the five trials included in the study, the authors of the Cochrane review were able to perform meta-analysis on only two (amounting to only 114 participants) to examine the incidence of the primary outcome. Of these two trials, one was rated as having a high risk of bias because of incomplete outcome data; the other was rated as having a low risk of bias overall. The three remaining trials were not included in the meta-analysis because outcomes were reported only at less than 1 month because of inadequate data. Two of the trials were performed at single centers, and the others, at multiple centers.Data regarding pain measurement were also lacking. The best validated pain scales, the visual analog scale or numerical descriptive scale, were not used in any of the trials. Four of the trials reported on changing pain intensity but used different methods of pain evaluation and could not be combined into the meta-analysis.
Although it was not the focus of the review, two of the trials suggested that steroids might have a significant effect on reducing acute pain and in accelerating healing of acute herpes zoster in the first month. The authors suggested that further studies to assess the effect of steroids on short-term pain and PHN should be performed.
Because of limited data and high risk of bias, it is difficult to draw any meaningful conclusion about the use of corticosteroids in reducing PHN after shingles. However, the existing limited evidence does not support the use of this treatment. Therefore, we assign a color rating of red (no benefit) to this treatment.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
See theNNT.com's previous reviews of this topic:
Corticosteroids for the Prevention of Post-Herpetic Neuralgia, November 26, 2010
Author
Daniel S. Kowalsky, MD; Allan B. Wolfson, MDPublished/Updated
March 15, 2019References:
Topical Antibiotics for Clinical and Microbiologic Cure of Bacterial Conjunctivitis
Benefits in NNT
9
NNT of 9 for clinical cure at 2-5 days
Harms in NNT
None were harmed
View As:
Source
Sheikh A, Hurwitz B, van Schayck CP, McLean S, Nurmatov U. Antibiotics versus placebo for acute bacterial conjunctivitis. Cochrane Database Syst Rev 2012;(9): CD001211.Study Population: 3,673 patients with bacterial conjunctivitis from 11 trials randomized to topical antibiotics vs. placebo
Efficacy Endpoints
Time to clinical and microbiologic cureHarm Endpoints
Adverse outcomes as reported in trials, for example, complications such as bacterial keratitis and orbital cellulitis; adverse reactions to antibiotics were not reportedNarrative
Acute bacterial conjunctivitis is an infective condition frequently resulting in mucopurulent ocular discharge, bulbar and palpebral injection, and discomfort. It may be difficult to differentiate between viral and bacterial conjunctivitis on clinical grounds, and swabbing eyes for cultures is not considered clinically practical. Therefore, although most cases are self-limited, antibiotics are typically given based on the belief that they decrease time to recovery, reduce sightthreatening complications, and reduce the rate of relapse.This review, an update of a previous Cochrane review from 2006, included 11 randomized control trials totaling 3,673 patients with bacterial conjunctivitis, whereas the prior review included five randomized control trials and 1,034 patients. The review included trials that made the diagnosis of bacterial conjunctivitis based on either clinical or microbiologic grounds.1 Clinical criteria required varied but generally included ocular discharge and conjunctival injection. Two of the trials required microbiologically proven bacterial conjunctivitis with the remainder making the diagnosis on clinical grounds. The primary outcomes of this review included both clinical and microbiologic cure rates.1 How cure was assessed varied between trials but, in general, it was defined by absence of symptoms or microbiologic eradication.
Data analysis from the trials indicated improved early (2- to 5-day) clinical cure rate of 40% (risk ratio [RR] = 1.36, 95% confidence interval [CI] = 1.15– 1.61) and microbiologic cure (RR = 1.55, 95% CI = 1.37–1.76). At 6 to 10 days (considered the “late” time point) antibiotics continued to show clinical benefit in clinical and microbiologic cure (RR = 1.21, 95% CI = 1.10–1.33; and RR = 1.37, 95% CI = 1.24–1.52 respectively). The absolute risk difference for early and late clinical cure were 11 and 9%, respectively, corresponding to NNTs of 9 and 11.1
Among subjects in the placebo groups, 30% achieved clinical cure by day 5, and 41% of cases had resolved by days 6 to 10. No serious outcomes were reported in either placebo groups or treatment groups.1
Caveats
Of the 11 included trials, two primary care-based trials were judged by the reviewers to be of high quality, with the remainder graded as being of poor quality. Nine of the 11 studies were judged to have a high risk of bias. Two of the 11 trials were done at primary care sites, and the remainder were performed at specialty care sites, suggesting the possibility of referral bias.1Interestingly, the natural history of bacterial conjunctivitis could not be inferred from the trials, as some of the trials used placebo eye drops containing an antiseptic that when applied three to four times a day was likely to have some clinical effect. Moreover, all of the included studies utilized different antibiotic regimens, which was a major contributor to the high degree of heterogeneity of the trials. Of note, the majority of the more recent trials utilized fluoroquinolones. Other factors contributing to heterogeneity included patient age, method of diagnosis, and definition of outcome measures. There was no recommendation regarding which antibiotic or duration of treatment was superior. Also of note, only two of the trials required microbiologic evidence to make the diagnosis of bacterial conjunctivitis, while the remainder allowed for bacterial conjunctivitis to be diagnosed clinically. This is a potential limitation of the study as it is possible that other forms of conjunctivitis were being treated.
In this review, 30% of the placebo groups achieved clinical cure by day 5, and 41% had resolved at 6 to 10 days. This suggests that the benefits of antibiotics were reflected in the rate of resolution of conjunctivitis but not in a reduction in complications, since no serious outcomes were reported in treatment or placebo groups.1 Given that complications such as orbital cellulitis are rare, however, a larger trial would be necessary to assess the efficacy of antibiotics in terms of reduction in complications.
In conclusion, despite the limited existing evidence (mostly poor quality with high risk of bias) the demonstration of consistent positive outcomes supports the use of topical antibiotics to treat bacterial conjunctivitis. The risk of adverse events associated with this treatment appear to be minimal. Therefore, we have assigned a color rating of green (benefits > harms) to this treatment.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Daniel S. Kowalsky, MD; Allan B. Wolfson, MDSupervising Editors: James McCormack, MD; Kabir Yadav, MD
See TheNNT.com's previous review of this topic: Topical Antibiotics for Bacterial Conjunctivitis, November 7, 2010
Published/Updated
References:
Antiviral Medications for the Prevention of Postherpetic Neuralgia After Herpes Zoster Infection
Benefits in NNT
None were helped
Harms in NNT
None were harmed
View As:
Source
Chen N, Li Q, Yang J, Zhou M, He L. Antiviral treatment for preventing postherpetic neuralgia. Cochrane Database Syst Rev 2014;(2):CD006866.Study Population: 1,211 patients with herpes zoster (shingles) infection randomized to treatment with antiviral medication or placebo in six trials
Efficacy Endpoints
Incidence of postherpetic neuralgia at 6 months after a first attack of shinglesHarm Endpoints
“Serious” (life-threatening, those leading to death) and “nonserious” adverse events during or within 2 weeks of treatmentNarrative
Postherpetic neuralgia (PHN) is a condition of persistent, refractory pain in an area previously affected by an acute herpes zoster infection. Age remains an important risk factor for the development of PHN, with 40% of patients older than 50 years and 75% of patients 75 years and older developing PHN after an initial episode of shingles.1 Persistent pain can lead to significant long-term problems such as depression, altered activities of daily living, and anorexia.1 Prior systematic reviews have suggested that treatment with antivirals within 72 hours of the onset of rash may reduce the incidence or duration of PHN.1The Cochrane systematic review discussed here is an update of a previous Cochrane review from 2009 and draws no new conclusions compared to the earlier review. The current review included six double-blind randomized placebo-controlled trials and a total of 1,211 patients. Five of these trials evaluated oral acyclovir, and the sixth trial evaluated famciclovir. Fifteen other studies were excluded for reasons such as a short follow-up interval, lack of placebo control, or initiation of treatment beyond 72 hours from the onset of rash.
This review found no significant difference between acyclovir and placebo in the incidence of PHN at 4 months (risk ratio [RR] = 0.75, 95% confidence interval [CI] = 0.51–1.11) or at 6 months (RR = 1.05, 95% CI = 0.87–1.27). The single study that evaluated famciclovir also failed to show reduced incidence compared to placebo. One trial comparing placebo and acyclovir with 46 participants reported statistically significant lower mean pain scores between 2 and 6 months using the visual analog scale–validated pain scale. The most common adverse drug events included nausea, vomiting, and headache, but these were not significantly different than in patients who received placebo.
The Cochrane review concludes that there is high-quality evidence that acyclovir does not reduce the incidence of PHN and suggests that further trials should focus on famciclovir and other antiviral agents since there is currently insufficient evidence to determine their efficacy.
Caveats
The results of prior studies may have been affected by how PHN was defined. This review defined PHN as pain persisting at least 120 days from the onset of rash. Four trials recorded data on herpetic neuralgia 1 month after rash onset, and this was analyzed in the review as an additional outcome measure not specified in the protocol. It found a statistically significant reduction in pain in the acyclovir group at 1 month after rash onset when compared to placebo (153/347 [44.1%] and 184/345 [53.3%], respectively; RR = 0.83, 95% CI = 0.71–0.96; p = 0.01).Only a few trials have examined pain and quality of life. Then Cochrane review suggests that these endpoints may be more useful measures in determining treatment efficacy rather than focusing only on the presence of symptoms. Only one of the trials included in this review was rated to be of good quality (based on design and avoidance of biases); the remaining studies were rated as fair because of unclear risk of bias. Biases identified included problems with blinding, allocation concealment, and random sequence generation. It appears that future research should adopt improved methods of data reporting so as to increase the likelihood of arriving at more clinically useful conclusions.
In conclusion, the existing evidence does not support the use of antiviral medication to prevent postherpetic neuralgia at 6 months. There are some data that suggest the benefit of antivirals on acute symptoms of zoster, but since this review focuses on antivirals for PHN prevention we have assigned a color rating of red (no benefit) to this treatment.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
See theNNT.com's previous reviews of this topic:
Antivirals for Preventing Post-Herpetic Neuralgia, November 23, 2010
Author
Daniel S. Kowalsky, MD; Allan B. Wolfson, MDPublished/Updated
March 4, 2019References:
Lung-protective Ventilation for Acute Respiratory Distress Syndrome
Benefits in NNT
10
1 in 10 were helped (life saved at 28 days from hospitalization)
12
1 in 12 were helped (life saved at time of hospital discharge)
Harms in NNT
Not reported
View As:
Source
Petrucci N, De Feo C. Lung protective ventilation strategy for the acute respiratory distress syndrome. Cochrane Database Syst Rev 2013;(2):CD003844.Study Population: 1,297 patients with acute respiratory distress syndrome from six trials
Efficacy Endpoints
28-day mortality, hospital mortality, mortality at the end of the follow-up period for each trialHarm Endpoints
Hypercapnia, acidosis, barotraumaNarrative
Acute respiratory distress syndrome (ARDS) is a type of acute, diffuse, and inflammatory lung injury. The Berlin definition (2012)1 includes the following ARDS criteria: 1) onset within 1 week of a known clinical insult, 2) bilateral opacities consistent with pulmonary edema, 3) respiratory failure not fully explained by cardiac failure or fluid overload, and 4) ratio of partial pressure of arterial oxygen (PaO2) to fraction of inspired oxygen (FiO2) of less than 300 mm Hg at a positive end expiratory pressure (PEEP) of 5 cm H2O. The new definition also categorizes ARDS as being mild for PaO2/FiO2 ratio of 200 to 300, moderate for PaO2/FiO2 ratio of 100 to 200, or severe for PaO2/ FiO2 ratio of less than 100 on PEEP of 5 cm H2O. Sepsis is the most common etiology for ARDS.2 Studies done before the introduction of Sepsis-3 in 2016 (which made the use of the term severe sepsis obsolete) showed that two of three cases with severe sepsis enter the health care system through the emergency department (ED). Prevalence of ARDS among medical patients in the ED has been estimated to be about 9%.2, 3 ED-based studies suggest an ARDS progression rate after admission of 27.5% in patients with severe sepsis and septic shock.4 The early onset and rapid progression of ARDS after ED admission which results in worsened outcomes suggest that time spent and treatments provided in the ED could alter the course of ARDS.According to a prospective multicenter cohort study (LUNG SAFE trial 2016),5 which was conducted with the aim of assessing the burden of acute hypoxemic respiratory failure requiring ventilatory support with a specific focus on ARDS, reported a prevalence of 10.4% for ARDS among intensive care admissions and 23.4% among ventilated patients. In this trial, the rates of hospital mortality were 35, 40, and 46% for patients with mild, moderate, and severe ARDS, respectively. There is considerable evidence that progressive lung parenchymal injury is induced by excessive alveolar distension by large tidal volumes.5 Lung-protective strategy has been developed to reduce further damage to already injured lungs. Lung-protective strategy is often divided into three strategies that include: 1) low tidal volume (6 mL/kg), 2) plateau pressure (Pplat) <31 cmH2O, and 3) appropriate PEEP.
A Cochrane systematic review published in 2013 by Petrucci and De Feo6 examined lung-protective strategies of mechanical ventilation for ARDS. This systematic review included six randomized controlled trials comprising 1,297 patients and compared mechanical ventilation with a lower tidal volume (VT) of ≤7 mL/ kg (lung-protective ventilation) versus VT of 10 to 15 mL/kg (conventional ventilation). Lung-protective ventilation was associated with a significantly decreased 28-day mortality (relative risk [RR] = 0.74, 95% confidence interval [CI] = 0.61 to 0.88, absolute risk reduction [ARR] = 10%, number needed to treat [NNT] = 10). Hospital mortality was similarly reduced (RR = 0.80, 95% CI = 0.69 to 0.92, ARR = 8%, NNT = 12). Overall mortality at the end of the follow-up period for each trial did not reach statistical significance (RR = 0.86, 95% CI = 0.69 to 1.06). The follow-up period varied from hospital discharge to 180 days in the largest trial included in the study.6
Plateau pressure is defined as airway pressure during the end expiratory pause and roughly reflects the level of alveolar overdistension. The mortality benefit for lung-protective strategy was evident only when the control group received “higher” (>31 cm H2O) Pplat (RR = 0.74, 95% CI = 0.63 to 0.87). The mortality rate between the groups was not statistically different when control groups received a “lower” Pplat (31 cm H2O or less).6
The Cochrane analysis reported insufficient data to analyze secondary outcomes such as development of multiorgan failure, duration of mechanical ventilation and total duration of mechanical support, total duration of stay in intensive care unit and hospital, longterm mortality, long-term health-related quality of life, and long-term cognitive outcome. The only secondary outcome with sufficient data for analysis was duration of mechanical ventilation (three trials, 288 patients) which was not statistically different between the groups.
A subsequent 2013 Cochrane review7 assessed the benefits and harms of high versus low PEEP in patients with ARDS. The use of higher levels of PEEP is part of the lung-protective strategy aimed at reducing ventilator-induced lung injury. PEEP is a mechanical maneuver that exerts a positive pressure in the lung and is used primarily to correct the hypoxemia caused by alveolar hypoventilation. The optimal level of PEEP in patients with ARDS is still controversial. The Cochrane review7 analyzed seven randomized controlled trials, and the authors found no difference in in-hospital mortality for those who received mechanical ventilation with high versus low PEEP, although there was a trend toward decreased mortality. There was no significant difference between the two groups for the number of ventilator-free days, with the latter referring to the number of days between successful weaning from mechanical ventilation and day 28 after study enrollment. Higher PEEP was associated with improved oxygenation on Days 1, 3, and 7, without an increase in barotrauma risk, defined as the presence of pneumothorax on chest radiograph or chest tube insertions for known or suspected spontaneous pneumothorax.
A multilevel mediation analysis that analyzed individual data from 3,562 patients with ARDS enrolled in nine previously reported randomized trials of nine randomized controlled trials suggested that it is the driving pressure (DP = VT/CRS) that is most strongly associated with survival.8 Driving pressure looks at the change in pressure across the alveoli, focusing on the ratio of the patient’s target tidal volume and the lung compliance, and targets functional lung rather than predicted lung size. Patient survival was linked to a lower driving pressure, with VT and PEEP being linked to survival only if they led to reductions in driving pressure.8
Caveats
Despite the fact that lung-protective strategy is widely accepted as the only intervention that improves mortality in ARDS, its use in the ED has been found to be uncommon, and hence prolonged ED length of stay can result in iatrogenic lung injury from excessively high tidal volumes.2 Evidence demonstrates that potentially injurious ventilator practices are common in the ED,9, 10, 11, 12, 13 especially because ventilator-associated lung injury can occur shortly after the initiation of mechanical ventilation.9, 14, 15 Early lung-protective ventilation during vulnerable periods results in subsequent benefit even when delivered for short periods of time.9, 16, 17 A before–after study of mechanically ventilated patients in the ED conducted by Fuller et al.17 in 2017 showed that 1) lung-protective strategies can be effectively implemented in the ED; 2) the implementation of an ED-based lung-protective ventilator protocol resulted in early utilization of lung-protective strategies in the intensive care unit, which increased subsequent adherence to lung-protective ventilation in ARDS patients; and 3) the intervention was associated with a significant reduction in pulmonary complications, hospital mortality, and health care resource use.17The 2013 Cochrane review6 of lung-protective strategies with low tidal volumes was heavily influenced by two studies, the ARDS Network 200018 and the study by Villar et al.,19 which are the only trials that showed a mortality benefit.6 Additionally, different lengths of follow-up and higher plateau pressure in control arms in two of the trials makes the interpretation of the combined results and long-term mortality difficult. Except for the one trial that reached the target sample size, all other five trials were terminated early. None of the trials reported a long-term outcome follow-up.
Lowering the tidal volume might not be without harm. Low tidal volumes can result in severe hypercapnia and acidosis which can in turn lead to increased intracranial pressure, depressed myocardial contractility, pulmonary hypertension, and depressed renal blood flow.20 The issue of adverse effects of a lower tidal volume was not addressed in the trials included in the Cochrane review. Specifically, the impact of acidosis and hypercapnia on the development of organ failure was not clear.6
Although the test of heterogeneity in the Cochrane meta-analysis6 was not statistically significant, the authors of Cochrane report that certain “hidden” heterogeneity due to clinical differences between the trials should be factored in. They also warn the readers about the fact that most of the trials did not report protocols of concomitant treatments and associated diseases (that is ventilator-associated pneumonia).6
In conclusion, the existing evidence supports the use of lung-protective strategy with low tidal volumes (6 mL/kg) and low plateau pressure (<30 mm Hg) in patients with ARDS due to mortality benefits. Despite the possibility of underreported harms, the benefits are prominent enough to justify assigning a color recommendation of green (benefit > harm) to this strategy.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
See theNNT.com's previous reviews of this topic:
Lung protective Ventilation Strategy for Intubated Patients with ARDS (Acute Respiratory Distress Syndrome), November 7, 2010
Author
Maida Hafiz, MD; Jennifer Stahl, MDSupervising Editors: Shahriar Zahtabchi, MD; Jarone Lee, MDPublished/Updated
References:
Video Laryngoscopy vs. Direct Laryngoscopy
Benefits in NNT
14
NNT of 14 for preventing failed intubation
11
NNT of 11 for preventing postintubation hoarseness
37
NNT of 37 for preventing airway trauma
Harms in NNT
No identifiable harms (no difference in the rate of hypoxia, sore throat, or mortality between the two groups)
View As:
Source
Lewis SR, Butler AR, Parker J, Cook TM, Smith AF. Videolaryngoscopy versus direct laryngoscopy for adult patients requiring tracheal intubation. Cochrane Database Syst Rev 2016;11:CD011136.Study Population: 7,044 adult patients (16 years and older) who required tracheal intubation across 64 trials
Efficacy Endpoints
Failed intubations, first successful attempts, airway trauma, postintubation hoarseness, and improved visualizationHarm Endpoints
Hypoxia, sore throat, and mortalityNarrative
Tracheal intubation is a critical step in securing the patient’s airway in a variety of emergent and nonemergent settings. Difficulties and complications may arise with this procedure, and alternative laryngoscopes that use video technology have been designed to improve visibility when airway difficulty is predicted or encountered. These devices may be flexible or rigid in design for the purpose of assisting in intubations, especially expected difficult intubations. Video laryngoscopes have been advertised as being able to reduce difficulty, failure, trauma, and other complications compared with direct laryngoscopy. Rigid video laryngoscopy uses a blade to retract the soft tissues and transmits a video image to a screen attached to the end of the handle or to a monitor. This design enables a lighted view of the larynx without direct “line of sight” and is also referred to as indirect laryngoscopy. In the Cochrane review discussed here,1 video laryngoscopy is compared to direct laryngoscopy in the tracheal intubation of adult patients.The Cochrane review included randomized control trials of both parallel and crossover design. No simulation or manikin studies were included. Participants were aged 16 years and older who required tracheal intubation electively for scheduled surgery, as well as participants requiring emergent intubation in the emergency department (ED) or the intensive care unit (ICU). The included trials compared the use of a video laryngoscope (VLS) of any model versus direct laryngoscopy with a Macintosh blade.
Nine types of VLS designs were used in the 64 included studies: GlideScope, Pentax AWS, C-MAC (to include DCI laryngoscope), McGrath, X-lite, C-MAC D-blade, Airtraq, Truview EVO2, and CEL- 100. Most studies compared the use of GlideScope, Pentax AWS, C-MAC, and McGrath. Some designs of Airtraq and Truview EVO2 can be used with and without a camera attachment, so only those studies which used with a camera were included.
The meta-analysis showed statistically significant decrease in number of failed intubations when VLS was used (odds ratio [OR] = 0.35, 95 confidence intervals [CI] = 0.19–0.65, absolute risk difference [ARD] = 7%, number needed to treat [NNT] =14). However, the rate of successful first attempt intubation between the groups was not statistically significant (OR = 1.27, 95% CI = 0.77–2.09).
Subgroup analyses carried out by type of scope revealed no significant difference in the number of failed intubations when the GlideScope, Pentax, or McGrath were compared with the Macintosh blade. The result for failed intubation remained statistically significant in favor of the C-MAC device in this analysis. Another subgroup analysis was performed based on predicted or known difficulty airways. This subgroup analysis revealed that fewer failed intubations occurred when a VLS was used in predicted or known difficult airways (OR = 0.28, 95% CI = 0.15–0.55, ARD = 7%, NNT = 14, n = 830 patients).
The systematic review also demonstrated statistically significant reduction in likelihood of laryngeal/airway traumas (22 trials, OR = 0.68, 95% CI = 0.48–0.96, ARD = 3%, NNT = 37) and fewer incidences of postoperative hoarseness (six trials, OR = 0.57, 95% CI = 0.36–0.88, ARD = 9%, NNT 11) when a VLS device was used.
Additionally, the Cochrane review analyzed intubation difficulty scale (IDS) and airway visualization. IDS scores were recorded in seven studies with 0 representing no difficulty. VLS increased the likelihood of a reported intubation difficulty scores of 0. Airway visualization was evaluated using Cormack-Lehane views. Achieving a Cormack-Lehane grade 1 view was also more likely with VLS.
Caveats
The use of video laryngoscopy was not adequately explored in the emergency setting. Of 64 studies included in the meta-analysis, only three studies included participants requiring emergency intubation (one in the ICU, one in the ED, and one in an out-of-hospital setting). Therefore, the findings of this systematic review might not be generalizable to emergency settings.All studies were subject to a high level of bias due to the inability to blind personnel to the type of laryngoscope used with each participant. As a result, the Cochrane authors downgraded the evidence for each outcome by one level for study limitations. Failed intubation, proportion of successful first attempts, and sore throat outcomes were assessed to be moderate-quality evidence, whereas outcomes of hypoxia, serious respiratory complications, and mortality were downgraded to very-low-quality evidence for imprecision. Additionally, a large number of studies with substantial heterogeneity that reported time to tracheal intubation were downgraded to very-low-quality evidence.
Most studies used an experienced anesthetist to perform laryngoscopies. However, it was not always clear from the papers whether anesthetists had equivalent experience with both devices. In light of improved patient centered outcome and relative safety, we assign a color recommendation of green (benefits > harms) to this intervention.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Abdullah Bakhsh, MBBS, FAAEM; Michael Ritchie, MDSupervising Editors: Shahriar Zehtabchi, MD; Jarone Lee, MDPublished/Updated
References:
Chewing Gum for Reducing Post-Cesarean Section Ileus
Benefits in NNT
14-17
1 in 14 to 17 had decreased postoperative ileus
Harms in NNT
Not assessed
View As:
Source
Pereira Gomes Morais E, Riera R, Porfírio GJ, et al. Chewing gum for enhancing early recovery of bowel function after caesarean section. Cochrane Database Syst Rev. 2016; (10): CD011562.Ciardulli A, Saccone G, Di Mascio D, Caissutti C, Berghella V. Chewing gum improves postoperative recovery of gastrointestinal function after cesarean delivery: a systematic review and meta-analysis of randomized trials. J Matern Fetal Neonatal Med. 2018; 31(14): 1924-1932
Study Population: More than 3,000 women (17 trials) who had just had cesarean section, mostly from the Middle East and Asia.
Efficacy Endpoints
Primary: postoperative ileus, time to flatus; Secondary: length of stay, time to defecation, need for enema, need for antiemetic, patient satisfactionHarm Endpoints
Not assessedNarrative
Postoperative ileus complicates a significant percentage of surgeries. By delaying the normal return to gastrointestinal function, postoperative ileus may increase patient discomfort and has been shown to prolong hospitalization by five days, increasing total costs by almost $1.5 billion annually in the United States.1 Surgeons and medical teams have tried numerous pharmacologic and nonpharmacologic interventions for postoperative ileus. The perfect intervention would be physiologic, effective, safe, and inexpensive. Early reintroduction of diet, although ideal, is not tolerated by some patients. Chewing gum may trigger salivation and the same neurodigestive processes that lead to normal gastrointestinal function and could represent a viable alternative to early diet reintroduction. Other research has shown gum chewing can reduce time to flatus, time to defecation, length of hospital stay, and the time to tolerate a diet in postoperative gynecologic oncology patients.2Two studies examined the effectiveness of chewing gum to reduce post-cesarean section ileus.3, 4 One review included 17 randomized controlled trials with more than 3,000 patients, and examined time to flatus and rate of ileus as primary outcomes.3 For time to flatus, they found 13 studies including 2,399 women. On average, chewing gum reduced this metric by seven hours. Regarding rate of ileus, four studies with 1,139 women demonstrated a reduction in incidence from 11% to 5%, yielding a number needed to treat (NNT) of 17. Secondary outcomes showed a reduction in time to defecation, as well as duration of hospital stay. The need for pain control or antiemetics did not differ between intervention and control groups.3
The other review focused on time to flatus as the primary outcome.4 Among 2,459 women, time to flatus was reduced from 29.5 to 23.1 hours. Secondary outcomes demonstrated reductions in rate of ileus (11.4% to 4.6%), time to bowel sounds, time to defecation, time to hunger, as well as decreased nausea and vomiting episodes and higher patient satisfaction.
An earlier source study found a similar reduction in rate of ileus (7.4%, NNT = 14), time to flatus, and time to defecation.5 The study also found a significant decrease in need for antiemetics and pain medication, but without an effect on length of stay.
None of the meta-analyses were able to truly assess harms because these were not reported in the included trials. Chewing gum did appear to be well tolerated with only three of 925 women discontinuing treatment. Given the well-established safety of chewing gum, true adverse events are likely insubstantial. Theoretical harms include choking, dental pain/temporomandibular dysfunction, and distaste. A separate study of patients who underwent minimally invasive gynecologic surgery demonstrated no related adverse effects.6
Caveats
Overall, our update focused on two meta-analyses that included studies of fairly low quality. This was limited by a general inability to blind patients to chewing gum, but a number of studies also lacked blinding of observers, and certain trials had incomplete concealment.Additionally, the authors report standardized mean differences for most of the outcomes. Although this allows for combining studies using different outcome measures, it also requires study populations to be similar. Given the heterogeneity in the studies, this may not be true. Heterogeneity was largely due to differences in patient populations, specific interventions, variations in diet, and outcome definitions. Included source trials varied in the nature of the intervention (e.g., when introduced, for how long each day, total duration) and in specifics of the outcome (i.e., definition of ileus). The validity of one of the trials was further limited by the use of multiple primary outcomes.3
Most of the included trials were conducted at single centers in low-income countries across the Middle East and Asia. Therefore, the external validity in the United States, Europe, and other more developed countries is uncertain. However, there is no reason to believe demographic differences would affect the proposed mechanism of action of chewing gum in this population or limit the generalizability of the results.
In the end, all of the studies suggest consistently significant improvements in the less patient-oriented outcomes (rate of ileus, time to flatus).3, 4, 5, 7 The more patient-oriented outcomes have been inconsistently demonstrated. Length of hospital stay was found to be increased in some trials,3, 7 but decreased in at least one other study.5 Similarly, nausea and vomiting have been reported to be increased in some studies,4, 5 and decreased in others.3 Although the current body of literature suggests a need for higher-quality, more robust research, the simplicity, frugality, and safety of chewing gum makes it a viable option to offer women undergoing cesarean section.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
See theNNT.com's previous reviews of this topic:
Chewing Gum for Reducing Post-Caesarian Section Ileus, January 6, 2015
Author
Gary Green, MDPublished/Updated
References:
Oxygen Therapy for Patients with Acute Myocardial Infarction
Benefits in NNT
Not applicable
Harms in NNT
No harms reported
View As:
Source
Sepehrvand N, James SK, Stub D, Khoshnood A, Ezekowitz JA, Hofmann R. Effects of supplemental oxygen therapy in patients with suspected acute myocardial infarction: a meta-analysis of randomised clinical trials. Heart. 2018;104:1691-1698.Study Population: Roughly 8000 subjects experiencing a heart attack
Efficacy Endpoints
Mortality, subsequent heart failureHarm Endpoints
Not assessedNarrative
Myocardial infarction (MI), or heart attack, is the death or injury of heart muscle when blood flow is abruptly cut off, and is a leading cause of death internationally with approximately 1.5 million cases per year in the United States.1 Supplemental oxygen in patients with MI (regardless of baseline oxygen level) is often used to improve oxygen supply to heart muscle, in hopes of reducing cardiac damage and thus improving outcomes.2, 3, 4The systematic review discussed here, including nearly 8000 subjects from 8 randomized trials,5 is dominated by a single nationwide registry trial of 6629 subjects in Sweden.6 In this study, patients with suspected myocardial infarction (based on either ischemic electrocardiographic or blood troponin changes) and normal oxygen saturation were randomly assigned to receive either supplemental oxygen by face mask for 6-12 hours or ambient air, in a non-blinded fashion.6 Oxygen therapy did not reduce death or any other endpoint measured at 2.1 years, and no subgroups were differentially benefited or harmed.6 These findings echoed the systematic review.5
Caveats
The main limitation of these data is inherent to the design of large registry trials like the Swedish one reported here. Registry trials are often pragmatic, eschewing steps such as blinding, and other features designed to minimize variation and bias. However, such trials have the advantage of being performed in ways that are consistent with usual practice, suggesting their results will translate more easily when implemented. The investigators in the Swedish trial did attempt to limit biases by, for instance, blinding outcome assessors to group assignments, and it is reassuring that the findings of 7 other trials included with in the systematic review5 are consistent with this large study. However, it remains feasible that methodologically more rigorous studies could find small outcome differences not detected in these data.In conclusion, existing evidence does not support routine supplemental oxygen for patients with acute myocardial infarction. Therefore, we have assigned a color recommendation of Red (No Benefit) to this intervention.
Author
Jia Jian Li, MDSupervising editors: Shahriar Zehtabchi, MDPublished/Updated
February 1, 2019References:
Thrombolytic Therapy for Pulmonary Embolism
Benefits in NNT
34
NNT of 34 for preventing one death
50
NNT of 50 for preventing one recurrent PE
Harms in NNT
10
NNH of 10 for one case of major hemorrhage
4
NNH of 4 for one case of minor hemorrhage
View As:
Source
Hao Q, Dong BG, Yue J, Wu T, Liu GJ. Thrombolytic therapy for pulmonary embolism (Review). Cochrane Database Syst Rev 2015;12:CD004437.Study Population: 2167 patients with pulmonary embolism from 17 randomized controlled trials
Efficacy Endpoints
Mortality, recurrent PEHarm Endpoints
Major or minor hemorrhageNarrative
Pulmonary embolism (PE) is a potentially life-threatening condition caused by occlusion in the pulmonary arterial circulation. PE can be categorized as low-risk, submassive (associated with evidence of right heart strain), or massive (associated with hemodynamic instability).1, 2, 3 The latter two subtypes are associated with increased morbidity and mortality.1, 2, 3 Traditionally, anticoagulation has been the mainstay of treatment. In massive PE, however, it is important to restore pulmonary blood flow rapidly, which is often accomplished by the use of thrombolytic agents or surgical embolectomy.1, 3 The medical literature has reported improvement in clot lysis, restoration of normal pulmonary circulation, a decrease in right heart strain, and improvement in long-term cardiac output and exercise tolerance with the use of thrombolytics.4, 5, 6 However, not all studies have demonstrated improvement in survival or other patient outcomes with thrombolytics, and thrombolytics carry an increased risk of bleeding.4, 7, 8, 9, 10, 11 The Cochrane review discussed here evaluates the efficacy of thrombolytics versus heparin alone for acute PE.12This Cochrane review is the third update of an original review published in 2006.13 The authors have included 17 randomized controlled trials (RCT), which included a total of 2167 patients who were diagnosed with acute PE confirmed by imaging or other validated assessment tool. The intervention group received thrombolytic therapy followed by heparin; the control group received heparin alone in 11 studies and heparin plus placebo in the remaining 6 studies. The primary outcomes included overall mortality, recurrent PE, major hemorrhagic events (defined as intracranial or retroperitoneal hemorrhage, decreased hemoglobin by more than 2 g/dL, or transfusion of 2 or more units of blood), and minor hemorrhagic events (defined as bleeding not meeting criteria for major bleeding). The included studies used several different types of thombolytics: alteplase (5 trials), streptokinase (5 trials), rt-PA (3 trials), urokinase (2 trials), tenecteplase (3 trials), and catheter-directed thrombolysis (1 trial). Eleven of the trials included patients with submassive PE, one trial evaluated massive PE, and 6 trials did not report the type of PE. Four trials included both massive PE and PE of unknown type. The sample sizes of the included trials ranged from 8 to 1006 patients, with the majority of RCTs including less than 100 patients. Mean age of included patients approximated 60 years.
This updated review found that thrombolytics plus heparin reduced the risk of death compared to heparin alone (odds ratio [OR]: 0.57; 95% confidence interval [CI] 0.37-0.87; absolute risk difference [ARD]: 2.9%; Number-needed-to-treat [NNT]: 34; low quality evidence). However, this effect was not significant when the four studies at high risk of bias were excluded (OR: 0.66; 95% CI 0.42-1.06). Thrombolytics also reduced the risk of recurrent PE (OR: 0.51; 95% CI 0.29-0.89; ARD: 2%; NNT: 50). Patients who received thrombolytics had a higher risk of major hemorrhagic events (OR: 2.90; 95% CI 1.95-4.31; ARD: 10.3%, Number-needed-to harm [NNH]: 10; low quality evidence) as well as minor hemorrhagic events (OR: 3.09; 95% CI 1.58-6.06; ARD: 25%; NNH: 4; low quality evidence).
Caveats
The meta-analysis discussed here has several important limitations. First, the overall evidence quality was low to very low due to limitations in trial design, influence of pharmaceutical companies, and small sample sizes. The mortality benefit was not present in the sensitivity analysis after exclusion of the four trials with unclear or high risk of bias. Studies used a variety of follow-up periods and thrombolytic agents, thus introducing significant heterogeneity. Sample sizes varied significantly as well, and most studies included less than 100 patients. No studies evaluated surgical embolectomy compared to thrombolytics. Randomization and blinding varied significantly among included trials. Four trials received funding from pharmaceutical companies.Identifying hemodynamically stable patients and unstable patients (massive PE) is imperative in PE. Among the trials included in the Cochrane analysis, however, only one trial, with a sample size of 8, specifically enrolled patients with massive PE.14 The impact of thrombolytics in patients with massive PE could not be determined in this review; however, the American College of Chest Physicians provides a grade 2C recommendation to administer thrombolytics in patients with acute PE and hypotension.15
A prior meta-analysis by Chatterjee et al. found decreased mortality with thrombolytic therapy.4 However, that review included several studies at high risk of bias and did not conduct sensitivity analyses.
Finally, the trials included in the Cochrane meta-analysis used a variety of thrombolytic agents, some of which (e.g. streptokinase) are no longer used because of the higher risk of hemorrhage.
In summary, low-quality evidence indicates that thrombolytics may reduce mortality and recurrent PE in patients with acute PE. However, thrombolytics significantly increase the risk of major and minor hemorrhagic events. Therefore, we assign a color rating of Yellow (unknown benefit) to thrombolytic therapy for undifferentiated PE. Further high-quality studies are needed to determine whether specific patient groups, particularly those with submassive or massive PE, may benefit from thrombolytic therapy or suffer harm from these agents.
Author
Brit Long, MD; Alex Koyfman, MD; Michael Gottlieb, MD, RDMSSupervising editors: Shahriar Zehtabchi, MD; Allan Wolfson, MDPublished/Updated
References:
Early Endovascular Thrombectomy for Large-vessel Ischemic Stroke Reduces Disability at 90 Days
Benefits in NNT
2.6
1 in 2.6 were helped (less disabled at 90 days)
5
1 in 5 were helped (achieved functional independence at 90 days)
Harms in NNT
Not applicable (the rates of symptomatic intracerebral hemorrhage, parenchymal hematoma type 2, and mortality did not significantly differ between the endovascular thrombectomy group and the control group)
View As:
Source
Goyal M, Menon BK, Van Zwam WH, et al. Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. Lancet 2016;387:1723–31.Study Population: 1,287 stroke patients from five trials (MR CLEAN, ESCAPE, SWIFT PRIME, REVASCAT, and EXTEND IA) with anterior circulation large-vessel occlusion (predominately middle cerebral artery) randomized to endovascular thrombectomy or standard medical therapy within 12 hours of symptom onset.
Efficacy Endpoints
Primary outcome: degree of disability on the modified Rankin Scale (mRS) at 90 days Secondary outcomes: Proportion of patients with functional independence (mRS 0–2) at 90 daysHarm Endpoints
Symptomatic intracranial hemorrhage, parenchymal hematoma type 2 (blood clot occupying > 30% of the infarcted territory with substantial mass effect), mortality within 90 daysNarrative
Anterior circulation large-vessel occlusion (LVO) of the internal carotid or middle cerebral artery is one of the most devastating ischemic stroke subtypes. Prior to 2015, evidence supporting endovascular thrombectomy for acute ischemic stroke was limited.1, 2 This was due to multiple factors, including low recanalization rates with previous-generation thrombectomy devices, inadequate neuroimaging inclusion criteria (patients enrolled in trials lacked target LVO and/or had large preexisting core infarcts), and selection bias—patients considered most likely to benefit from thrombectomy underwent the procedure outside of clinical trials.1, 2 However, five randomized controlled trials3, 4, 5, 6, 7 published in 2015 established that endovascular thrombectomy significantly reduces disability in acute ischemic stroke when performed with stent retrievers in the setting of anterior circulation LVO and minimal core infarct burden. Four of these five trials were terminated early due to overwhelming treatment benefit, limiting analysis of secondary outcomes. A patient-level meta-analysis of all five trials was published in The Lancet in 2016.8 All subjects included in the meta-analysis had an anterior circulation LVO stroke. The median (interquartile range [IQR]) age was 68 (57–77) years with median (IQR) National Institutes of Health Stroke Scale (NIHSS) 17 (14–20) and Alberta Stroke Program Early CT Score (ASPECTS) 9(7–10). Median (IQR) time to recanalization of the occluded vessel with endovascular thrombectomy was 285 (210–362) minutes.The aggregate clinical trial data strongly favor endovascular thrombectomy over medical management for anterior circulation LVO with a number needed to treat (NNT) of 2.6 for one additional patient to achieve improved functional outcome, defined as improvement by at least one level on the modified Rankin Scale (mRS) at 90 days (adjusted odds ratio [aOR] = 2.49, 95% confidence interval [CI] = 1.79–3.53; please refer to Figure 1A for the mRS). Additionally, for every five patients treated with endovascular thrombectomy, one additional patient achieved functional independence (mRS 0–2) at 90 days (46%vs. 26.5%, absolute risk difference = 19.5%, aOR = 2.71, 95% CI = 2.07–3.55; Figures 1B and 1C). These findings were consistent across all subgroups (age, sex, NIHSS, ASPECTS, site of intracranial occlusion, intravenous tPA eligible or ineligible, and time from onset to randomization), suggesting that the benefit of thrombectomy is generalizable to a broad spectrum of patients with anterior circulation LVO.8 Importantly, this strengthens the evidence that endovascular therapy should not be withheld on the basis of factors such as age or tPA eligibility.8
Caveats
All five clinical trials were conducted at experienced, high-volume tertiary stroke centers. Therefore, these results may not be replicated at clinical sites without similar levels of stroke and neurointerventional expertise. In addition, these results cannot be extrapolated to all stroke patients. The benefit of endovascular treatment for stroke patients with large infarcts (representing irreversible ischemic injury), but substantial viable ischemic tissue remains to be determined. Additional stroke subgroups for which thrombectomy is yet to be definitively studied include posterior circulation stroke, distal vessel occlusion, patients with minor deficits, and those with premorbid disability. Notably, two recent clinical trials have demonstrated a benefit for delayed thrombectomy (6–24 hours from stroke onset) in patients selected using more stringent clinical and advanced neuroimaging criteria.9, 10 These highly selected patients represent a more homogenous stroke population distinct from the trials in the meta-analysis of Goyal et al.8There was no significant difference in symptomatic intracerebral hemorrhage (aOR = 1.07, 95% CI = 0.62–1.84), parenchymal hematoma type 2 (aOR = 1.04, 95% CI = 0.63–1.72), and mortality (aOR = 0.73, 95% CI = 0.47–1.13) between the thrombectomy group and the medical group.8 One plausible explanation for this finding is that, despite the associated procedural risk, thrombectomy patients ultimately have smaller ischemic stroke volumes than nonthrombectomy patients leading to lower rates of postprocedural spontaneous hemorrhagic transformation.6
The mRS, which is commonly used in stroke clinical trials, is a discrete functional outcome score with high inter-rater reliability and little room for subjective interpretation.11 Although participants and their families were not blinded to the treatment arm, to minimize bias, each individual trial employed a PROBE (prospective randomized open blinded endpoint) design, in which all outcome assessors remain blinded. Additionally, all outcomes were adjudicated by study personnel certified in scoring the mRS.
In acute stroke trials, functional outcome can be analyzed by binary analysis (dichotomized ordinal outcome scales, i.e., “good” vs. “poor”) or by shift analysis (which evaluate outcomes over the entire scale range). One limitation of the mRS shift analysis employed in these trials is that it values each mRS strata equally. However, studies have shown, for example, that most people attach a higher value to an improvement from mRS 3 to 2 than to an improvement from mRS 1 to 0 (Figure 1A). This was accounted for in the DAWN trial,9 which employed a weighted shift analysis as one of the co-primary endpoints. Despite this limitation, the shift analysis has become convention in stroke trials because it provides information across a wider range of outcomes and, hence, is considerably more clinically relevant than traditional dichotomized outcomes.
In conclusion, we assigned a color recommendation of green (benefit > harm) to this intervention because of evidence of patient-centered benefit for early endovascular thrombectomy in anterior circulation LVO stroke and absence of significant harm. This benefit was evident across a wide range of ages and was present irrespective of tPA eligibility.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Mona Al Banna, MB, BCh, BAO, MSc; Christopher D. Streib, MD, MSSupervising Editor: Shahriar Zehtabchi, MD
Published/Updated
January 11, 2019References:
Tissue Plasminogen Activator (tPA) For Acute Ischemic Stroke
Benefits in NNT
The NNT for ischemic stroke treatment to achieve one additional patient with excellent functional outcome (mRS 0-1) is time-dependent and is displayed below by onset to treatment time
10
0-3h: NNT of 10
19
3-4.5h: NNT of 19
50
4.5-6h: NNT of 50
Harms in NNT
Fatal intracranial hemorrhage within 7 days of treatment
40
0-3h: NNH of 40
50
3-4.5h: NNH of 50
40
4.5-6h: NNH of 40
71
90-day all-cause mortality: NNH of 71
View As:
Source
Emberson J, Lees KR, Lyden P, et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: a meta-analysis of individual patient data from randomised trials. Lancet 2014; 384(9958): 1929-35.Study Population: 6756 patients in nine randomized trials comparing alteplase with placebo or open control for the treatment acute ischemic stroke. Trials of other thrombolytic agents (e.g. streptokinase, urokinase) were excluded.
Efficacy Endpoints
Excellent functional outcome defined as a modified Rankin Scale (mRS) score of 0-1 assessed at 90-180 days after stroke. The mRS is an inclusive efficacy and harm outcome because it includes harm related to poor outcomes.Harm Endpoints
Fatal intracerebral hemorrhage, all-cause mortality. (Note that harm endpoints are included in the efficacy endpoint because the mRS includes all-cause mortality as a poor outcome)Narrative
Stroke is a devastating condition that leaves most survivors with permanent neurological disability. Worldwide, stroke is the second leading cause of death and has a declining age-adjusted incidence in high-income countries but a rapidly rising incidence and impact in lower- and middle-income countries. Ischemic stroke is the most common stroke type, accounting for roughly 85% of all strokes. Ischemic stroke is caused by an acute occlusion of an intracranial artery or one of four extracranial cervical arteries leading to the brain. Treatment with thrombolytic drugs targets the occlusion and is designed to restore blood flow to the brain. Thrombolytic drugs are effective in recanalizing occluded brain arteries only some of the time. Large proximal artery occlusions (e.g. carotid artery or middle cerebral artery stem) are opened 10-35% of the time, and smaller more distal arteries, up to 60-80% of the time. Large proximal artery occlusions are best treated with a combination of thrombolysis and endovascular thrombectomy.1, 2Initial thrombolytic trials, which assessed streptokinase, were uniformly negative; streptokinase was shown to be harmful, resulting in a higher rate of fatal intracranial hemorrhage and overall greater mortality compared to placebo. As a consequence, streptokinase has been abandoned and should no longer be considered for stroke. Individual trials of alteplase have been both positive and neutral. Treatment efficacy is highly time-dependent, while treatment harm is independent of time, within a 6-hour treatment window. The therapeutic index (i.e, benefit vs harm) is therefore much larger when treatment is administered quickly after stroke onset, and much smaller when given at later times. More recent trials have examined comparisons of alteplase dose (0.6mg/kg vs. 0.9mg/kg) and the use of advanced brain imaging to select patients for treatment who fall outside the usual time-based rules.3, 4, 5
Stroke outcomes are assessed using a global outcome which includes both efficacy (good neurological functional outcome) and harm (death, fatal ICH and ICH leading to poor functional neurological status) outcomes in a single composite measure. The modified Rankin Scale assesses neurologic functional outcome on a 7-point scale from 0 (no symptoms at all) to 5 (bedbound) and 6 (death). Outcome is typically assessed at 90-180 days after stroke onset. The outcome is validated and reproducible. The score is commonly characterized and evaluated statistically as follows: mRS 0-1 = excellent functional outcome; mRS 0-2 = independent functional outcome; mRS 3-6 = dependency or death.
This review summarizes the pooled individual-patient meta-analysis of randomized phase-3 trials comparing alteplase to placebo, comprising 6756 patients.6 Assessments of streptokinase or other thrombolytic agents are not included because they are known to be harmful. The treatment benefit is most pronounced when treatment is delivered rapidly after stroke onset (NNT = 10 for treatment within 3 hours of stroke onset). Beyond 4.5 hours from stroke onset, there is no average benefit. With increasing awareness of the importance of rapid evaluation and treatment and the establishment of acute stroke teams that include emergency physicians and specialist stroke physicians, door-to-treatment times of under 30 minutes can be routinely achieved. Treatment of ischemic stroke due to large vessel occlusion within 30 minutes of hospital arrival, as compared to >30 minutes after hospital arrival, is associated with a NNT of 5; for every 5 patients with large vessel occlusion treated with intravenous alteplase within 30 minutes of hospital arrival, one additional patients will have an independent functional outcome (mRS 0-2).
However, only one third of stroke patients achieve an excellent neurologic outcome with thrombolysis; thus, a majority still fare poorly with only alteplase treatment. [Note that the rising proportion of patients with a good outcome in the control group in later time windows reflects that fact that stroke severity is lower at later time points; more severe stroke presents early.] The combination of alteplase plus endovascular thrombectomy for severe strokes due to proximal large vessel occlusion improves the proportion of patients with independent functional outcome to 50%.1
Stroke treatment with alteplase is associated with an increased risk of fatal intracranial hemorrhage (2.7% vs. 0.4%) in the first 7 days (SITS-MOST criteria type 2 parenchymal hemorrhage - which defines clinical symptomatic and large radiological ICH). The absolute risk increase is 2.0-2.5% (NNH = 40-50).6 Additional non-fatal symptomatic hemorrhage in the first 7 days (SITS-MOST criteria type 2 parenchymal hemorrhage) is increased (1.0% vs 0.2%). The absolute increase is 0.8% (NNH = 125). When hemorrhage is defined radiologically only as PH-2 type hemorrhage (including all fatal, symptomatic and asymptomatic large parenchymal hematoma), there is an absolute increase of PH-2 type hemorrhage in the first 7 days of 5.5% (6.8% vs 1.3%). In addition, the natural history of ischemic stroke is to evolve some degree of asymptomatic (usually petechial) hemorrhage. Asymptomatic hemorrhage can be detected by magnetic resonance imaging in nearly all ischemic strokes, with and without thrombolysis treatment, and is not associated with poorer outcome. Poor outcomes (death or disability) due to hemorrhage are included in the global outcome assessment using the mRS. It is important not to conflate acute hemorrhage with poor outcome at 90 days. While acute hemorrhage is often serious and even fatal (~50% of symptomatic ICH is fatal), it is not necessarily the dominant cause of poorer long-term outcomes; overall, the major prevalent cause of poor outcomes is large ischemic stroke, which are reduced with thrombolysis. The NNH for all-cause mortality is 71.6
There is a tradeoff involved in stroke thrombolysis. Treatment, particularly fast treatment, results in a greater number of patients with independent neurologic functional outcome, but with a small risk of early death due to fatal intracranial hemorrhage.
Caveats
Over the two decades since the first ischemic stroke thrombolytic trials were reported, it has become clear that acute ischemic stroke treatment is best delivered by a team with expertise in emergency critical care, rapid neurological assessment, interpretation of brain imaging, and access to stroke unit care. It is within this kind of structure that rapid, expert treatment can be delivered, and in multiple hospitals around the world this approach has resulted in improved outcomes for stroke patients. Skepticism about thrombolytic treatment has arisen mainly in the emergency medicine literature. However, discussion of the strengths or limitations of the original trials is beyond the scope of this evidence-based summary. When treatment has been attempted in the absence of a collaborative team structure, trial results have not been duplicated. Without the necessary structure, real-world treatment has been either neutral or frankly harmful. To apply this therapy well and duplicate the results of published trials requires the support of a dedicated stroke team. We encourage dialogue and teamwork between stroke neurologists and emergency physicians.A limitation of meta-analyses is the selection criteria for the included trials. An advantage of pooling individual patient data is that a time-effect can be estimated. Characteristics of the individual trials should be noted. The majority of data on treatment within the early time window (0-3h) originates from two parallel trials conducted in the United States only. Most data from the later-time window (3-6h) come from European trials. Few data are available on patients from Asia. All but one trial used a dose of 0.9 mg/kg alteplase. Four of the trials were exclusively funded by industry; five were funded by public granting organizations. These results are corroborated by the Cochrane meta-analysis that examined trial-level outcomes.7
Treatment of ischemic stroke with intravenous alteplase is strongly recommended (Level 1 or Grade A) by all major stroke guidelines. However, it is an intervention that is best provided by a well-organized and expert stroke team comprising emergency physicians and specialist stroke physicians in a setting that can provide excellent subsequent care in a dedicated stroke unit.
Author
Michael D. Hill, MD; Noreen Kamal, PhD; Eddy Lang, MDSee theNNT.com's previous reviews of this topic: Thrombolytics for Acute Ischemic Stroke, March 25, 2013
Published/Updated
References:
"” "” "” "” "” "” "”
HPV vaccines for prevention of cervical pre-cancer in adolescent girls and women
Benefits in NNT
60
NNT of 60 for preventing any cervical pre-cancer
Harms in NNT
Not applicable: No significant difference between the vaccinated and unvaccinated individuals
View As:
Source
Arbyn M, Xu L, Simoens C, Martin-Hirsch PPL. Prophylactic vaccination against human papilloma viruses to prevent cervical cancer and its precursors. Cochrane Database of Systematic Reviews 2018, Issue 5. Art. No.: CD009069. DOI: 10.1002/14651858.CD009069.Population: 73,428 mostly between 15 to 25 years of age from 26 trials
Efficacy Endpoints
Pre-cancer cervical lesions, cervical cancerHarm Endpoints
Serious adverse events, death, pregnancy-related adverse events (spontaneous abortion, stillbirth, congenital malformations)Narrative
Human papillomaviruses (HPV) are sexually transmitted infections that are common in young people. In the United States, HPV infects nearly 80 million people. Approximately, one in four Americans are currently infected with HPV. About 14 million individuals become infected with HPV each year in the United States.1 Usually these viruses are cleared by the immune system and pose little threat. However, high-risk (hr) types of HPV can cause persistent infection which can lead to precancerous changes in cells. Precancerous changes are defined as infection of at least two thirds of the surface layer of cervical cells. Some people with this pre-cancerous abnormality will go on to develop cervical cancer; unfortunately, we don’t know which individuals with infection will clear these changes and which will develop cancer. A number of different hrHPV types cause cervical pre-cancer and cancer, the most important of which are HPV16 and 18. These high-risk types cause about 70% of cervical cancers worldwide.2 Cervical cancer is the fourth most common cause of cancer in women worldwide, and 86% of worldwide cervical cancer cases occur in developing countries. The lifetime risk of cervical cancer is approximately 0.6% and the chance of dying from cervical cancer is approximately 0.2%.3 Vaccination with HPV-like particles triggers the production of antibodies which protect against the development of precancerous cervical changes. The Cochrane review discussed here assesses the benefits and harms of prophylactic HPV vaccination against cervical pre-cancer and HPV16/18 infections in adolescent girls and women.2 The assumption is that if HPV vaccine effectively prevents precancerous changes by hrHPV infection, vaccination should prevent a majority of cases of cervical cancer.A total of 26 studies involving 73,428 adolescent girls and women were included in the Cochrane meta-analysis. The authors performed pre-planned analyses separately in groups of women 15-25 and 26-45, as the majority of trials enrolled subjects 25 years of age or younger.2 Participants were divided into sub-groups; the first group had no evidence of baseline infection with high-risk human papilloma viruses types, the second group had no evidence of baseline infection with HPV types included in the vaccines, and the third group had unknown baseline infection with HPV. The review included all trials that evaluated vaccine safety over a period 0.5 to 7 years and ten trials with follow-up 3.5 to 8 years. Cervical cancer could not be evaluated as an outcome due to inadequate study sample sizes and insufficient follow up duration.2 The summary of the Cochrane review analyses results is presented in the table.
Table. Efficacy of HPV vaccine for preventing cervical pre-cancer in different subgroups based on age and type of cervical abnormality
| Subgroups of women aged 15-25 | Cervical pre-cancer* with HPV 16/18 | Any cervical pre-cancer |
| Women hrHPV negative | ARD: 1.6% NNT: 63 RR: 0.01 (0 to 0.05) Quality of Evidence: High | ARD: 1.8% NNT: 56 RR: 0.37 (0.25 to 0.55) Quality of Evidence: High |
| Women HPV 16/18 negative | ARD: 1.07% NNT: 94 RR: 0.05 (0.03 to 0.10) Quality of Evidence: High | ARD: 1.36% NNT: 74 RR 0.41 (0.32 to 0.52) Quality of Evidence: High |
| All Women with or without HPV infection | ARD: 1.84% NNT: 55 RR: 0.46 (0.37 to 0.57) Quality of Evidence: High | ARD: 1.68% NNT: 60 RR 0.70 (0.58 to 0.85) Quality of Evidence: High |
| Subgroups of women aged 25 to 45 | ||
| Women free of hrHPV | No data | No data |
| Women free of HPV 16/18 | No data | No data |
| All Women with or without HPV infection | RR: 0.74 (0.52 to 1.05) Difference not significant Quality of Evidence: moderate | RR: 1.04 (0.83 to 1.30) Difference not significant Quality of Evidence: moderate |
Abbreviations: hr: High risk; HPV: human papilloma virus; NNT: Number-needed-to-treat, ARD: Absolute risk difference; RR: Relative risk
* Cervical intraepithelial neoplasia, grade 2 and higher (CIN2+). Data for CIN3+ are not reported.
There were no significant differences in serious adverse events between vaccinated and unvaccinated individuals (RR: 0.98, 95% CI 0.92 to 1.05; 71,597 participants; high-quality evidence) independent of type or dosage of HPV vaccine. The HPV vaccine administration was not associated with increased risk of pregnancy-related adverse outcomes either (moderate to high quality evidence).
Caveats
Vaccination against HPV is performed to reduce the incidence of cervical cancer. Unfortunately, this outcome could not be assessed in the meta-analysis, because the included trials lacked sufficient power and adequate follow up.2 Additionally, this meta-analysis extracted efficacy data from peer-reviewed published reports. Incomplete reporting of the original trials limited the number of studies that could be included in each of the analyses.2 Furthermore, HPV vaccines were not yet standardized at the time of the studies so the potency for specific anti-HPV serological responses may be a variable.4The review identified several notable biases. Computed efficacy estimates for women who received only one or two doses were calculated instead of measured. Another post hoc analysis limitation was the variation caused by counting events for participants who received at least one dose at day one, compared to counting events for participants who received all three doses on the day of the last administration.2 Another source of bias originated from the fact that for several outcomes no information was available for mid-adult women. Finally, the comparison of the risks of adverse events was compromised by the use of different products administered to participants in the control group, varying from adjuvant (often aluminum hydroxide or other aluminum compound) or an alternative vaccine (often Hepatitis A or Hepatitis B),5 and therefore, the pooled risks of adverse effects associated with HPV vaccines and the assumed risks for control groups must be interpreted carefully. It must be noted that 25 out of the 26 trials were sponsored by the vaccine manufacturers. However, the analyses of bias performed by the Cochrane review found little evidence of important bias due to trial sponsorship.
The quality of the evidence was deemed to be high, since the data included more than 70,000 women from randomized trials.2 Heterogeneity was minimal in this review since data series did not combine participants with different baseline HPV status (presence of hrHPV DNA, presence of DNA of the HPV vaccine types). Moreover, younger (15 to 26 years) women were distinguished from mid-adult women (24 to 45 years). Efficacy estimates were not significantly different by vaccine type, and jointly pooled estimates were retained. Only when significant heterogeneity by vaccine types was noted, were separate efficacy estimates by vaccine type pooled.2 When significant heterogeneity persisted, the review used meta-regression to investigate sources of heterogeneity, such as serological status, study design items, study size and sexual history.6, 7 This process decreased any heterogeneity presented in the data.
While the reason for higher efficacy of HPV vaccine in younger women is not discussed in the Cochrane review, it is likely that younger age allows for more likelihood of exposure to HPV and potentially developing pre-cancerous cervical lesions. This factor could justify the recommendations that HPV vaccines should be administered as early as possible in adolescence. Finally, it is important to remember that the lifetime risk of cervical cancer is approximately 0.6%.3 So even if the vaccine is 100% effective at preventing cervical cancer, the best NNT for cancer prevention over a lifetime would be 167.
As of December 2018, the cost of HPV vaccine is approximately $261-$461.8
Conclusion: The evidence presented in this meta-analysis shows the HPV vaccination confers significant benefit in preventing cervical pre-cancer. The effect is higher for lesions associated with HPV16/18. The data also demonstrates an absence of serious adverse events. Therefore, we have assigned a color recommendation of Green (Benefit > Harm) to this vaccine.
The original manuscript was published in the Journal of Evidence-Based Healthcare as part of the partnership between TheNNT.com and the journal.
Author
Jia Jian Li and Jessica Stetz, MDSupervising Editor:Shahriar Zehtabchi, MD
Published/Updated
December 21, 2018References:
Early Invasive Management of Acute Coronary Syndromes
Benefits in NNT
No deaths were prevented
62
1 in 62 avoided a new heart attack in the next year
9
1 in 9 experienced less chest pain
15
1 in 15 avoided rehospitalization
Harms in NNT
39
1 in 39 experienced a heart attack during or immediately after the procedure
33
1 in 33 experienced a major bleeding episode during the procedure
View As:
Source
Fanning JP, Nyong J, Scott IA, Aroney CN, Walters DL. Routine invasive strategies versus selective invasive strategies for unstable angina and non-ST elevation myocardial infarction in the stent era. Cochrane Database Syst Rev. 2016;(5):CD004815.Study Population: Patients with unstable angina or acute non-ST-segment elevation myocardial infarction (NSTEMI)
Efficacy Endpoints
Death, myocardial infarction, angina symptoms, and rehospitalization at six- to 12-month follow-upHarm Endpoints
Bleeding, periprocedural myocardial infarction, deathNarrative
Reperfusion therapy for acute ST-segment elevation myocardial infarction (STEMI) has been shown to be beneficial. However, there is controversy regarding the management of unstable angina and NSTEMI. Fowler and Conti coined the term unstable angina in 1971 for patients who did not meet the criteria for acute myocardial infarction or stable angina.1, 2 The term may be outdated now with the increased sensitivity of cardiac troponins. Patients with unstable angina or patients in the “gray zone” of symptomatic ischemia can now be diagnosed as having NSTEMI.3 This Cochrane review4 updates the 2010 review5 of early invasive management for acute coronary syndrome that identifed five trials. That systematic review found a statistically significant reduction in myocardial infarction (2%) with the invasive strategy and concluded that an early invasive strategy was superior to a noninvasive strategy.5In this updated Cochrane review,4 the authors added three new trials with a total of 1,099 participants to the metaanalysis. Therefore, the updated Cochrane review represents eight randomized controlled trials with a total of 8,915 participants randomized to an invasive strategy, whereby all patients undergo coronary angiography and revascularization (as necessary), or a conservative strategy in which medical therapy is used initially and patients are selected for cardiac catheterization only if there is evidence of persistent myocardial ischemia. Patients included in the studies were at least 18 years of age, had an episode of chest pain at rest, and had at least one of the following criteria: (1) electrocardiography changes including new ST depression, transient ST elevation (less than 20 minutes), or ischemic T wave inversions in at least two contiguous leads; (2) elevated cardiac markers; or (3) known coronary artery disease. Patients were excluded if they had persistent ST elevation, secondary causes of acute myocardial ischemia or cardiac biomarker elevations, severe cardiogenic shock or congestive heart failure, arrhythmias that required catheterization, refractory symptoms, coronary revascularization within the past 30 days, or intolerance to anticoagulation or antiplatelet therapy.
This new Cochrane review concludes that an early invasive strategy does not provide a mortality benefit (relative risk [RR] = 0.87; 95% confidence interval [CI], 0.64 to 1.18).4 However, invasive strategy reduced the rate of refractory chest pain (absolute risk reduction [ARR] = 11%; RR = 0.64; 95% CI, 0.52 to 0.79) and the risk of nonfatal myocardial infarction within a year (ARR = 1.5%; RR = 0.79; 95% CI, 0.63 to 1.00) compared with patients who were managed conservatively. The rate of rehospitalization was also reduced with an early invasive strategy (ARR = 6.7%; RR = 0.77; 95% CI, 0.63 to 0.94).
More complications were noted in the invasive group. There was an increase in periprocedural myocardial infarction (absolute risk increase [ARI] = 2.5%; RR = 1.87; 95% CI, 1.47 to 2.37) in the invasive management group as well as an increase in the risk of major bleeding events (ARI = 3%; RR = 1.73; 95% CI, 1.3 to 2.31). Increased risks of periprocedural heart attack and major bleeding caused by the procedure appear to offset any benefits in preventing future heart attacks. However, patients might benefit from the procedure simply from reducing chest pain or preventing
rehospitalization.
Caveats
A limitation of the presented data is that when conservative therapy (ongoing pain or ischemia) was ineffective, patients underwent invasive angiography and stenting as necessary. This makes it difficult to assess whether invasive therapy is beneficial over conservative therapy alone. In addition, it introduces bias in the rates of rehospitalization. Patients who switch from conservative treatment to an invasive strategy are more likely to be rehospitalized.The authors of the systematic review deemed the trials to have a high risk of performance-blinding bias. All studies were deemed to have low risk of attrition bias.
The updated Cochrane review4 added two trials on patients with an increased risk of mortality (the Italian Elderly ACS6, 7 and LIPSIA-NSTEMI8 trials), which were not included in the 2010 Cochrane review.5 The participants in the Italian Elderly ACS trial were all at least 75 years of age, which has been associated with increased rates of adverse effects.6, 7 The LIPSIA-NSTEMI trial included participants who already had elevated troponin levels, and routine use of glycoprotein IIb/IIIa inhibitors may have contributed to increased adverse events with the invasive strategy.8 Inclusion of these high-risk populations may have caused the overall outcomes to be less favorable in both groups.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Sally Liang, MD; Michael Ritchie, MDPublished/Updated
References:
Conti CR, Greene B, Pitt B, et al. Coronary surgery in unstable angina pectoris [abstract]. Circulation. 1971;44(suppl II):II-154.
Vaccines for Preventing Influenza in Healthy Individuals
Benefits in NNT
71
NNT of 71 for preventing influenza
29, 5
NNT of 29 for elderly and 5 for children
29
NNT of 29 for preventing influenza-like illness
42, 12
NNT of 42 for elderly and 12 for children
Harms in NNT
125
1 in 125 experienced fever after vaccination
View As:
Source
Demicheli V, Jefferson T, Ferroni E, Rivetti A, Di Pietrantoni C. Vaccines for preventing influenza in healthy adults. Cochrane Database of Systematic Reviews 2018, Issue 2. Art. No.: CD001269Study Population: More than 80,000 healthy adults from 52 clinical trials aged 16 to 65 years, including pregnant women, over a single influenza season in North America, South America, and Europe who received vaccination between 1969 and 2009
Efficacy Endpoints
Incidence of influenza infection and influenza-like illnessHarm Endpoints
Adverse events including malaise, fever, arthralgia, rash, headache, and neurological harmsNarrative
Influenza is an acute respiratory infection that imposes a heavy burden on society.1 The illness itself usually lasts a few days but the residual symptoms of cough and malaise can last for weeks. In addition, it can cause complications such as otitis media, pneumonia, secondary bacterial pneumonia, exacerbations of chronic respiratory disease, bronchiolitis, febrile seizures, Reye’s syndrome, and myocarditis.1 Vaccines have been developed in attempt to minimize the effects of influenza. However, given the yearly antigenic changes of the virus, a new vaccine has to be developed, produced, and administered to the population every year.2The Cochrane review discussed here assesses the efficacy of vaccines in preventing influenza in healthy adults including pregnant women.2
A total of 52 clinical trials of over 80,000 healthy adults aged 16 to 65 years over a single influenza season in North America, South America, and Europe who received vaccination between 1969 and 2009 were included in this review. The results presented were from 25 studies comparing inactivated parenteral influenza vaccine against placebo control groups.2 Fifteen of the included trials were industry sponsored.2 The review concluded that healthy adults who received inactivated parenteral influenza vaccine rather than placebo were at lower risk of influenza infection; from 2.3% in the control group to 0.9% in the vaccinated group (number-needed-to-vaccinate [NNV]: 71; absolute risk difference [ARD] 1.4%; relative risk (RR): 0.41, 95% confidence interval [CI], 0.36-0.47, moderate certainty). People who received vaccination also experienced less influenza-like illness (ILI) (NNV: 29; RR: 0.84, 95% CI, 0.75-0.95). The rate of hospitalization and time out of work endpoints were not significantly different between vaccinated and un-vaccinated groups.2
There was an increase in risk of developing fever in the vaccinated group from 1.5% to 2.3% (RR: 1.55, 95% CI 1.26-1.91; ARD: 0.8%; Number-needed-to-harm [NNH]: 125).2 People also reported more nausea and vomiting after vaccination, however the risk of these adverse events was not statistically significant between the groups. No association was found between vaccination and Guillain-Barre syndrome or other neurological diseases.2 There was also no increased risk of abortion, congenital malformation, prematurity, or neonatal death in vaccinated pregnant women.2
Two other Cochrane systematic reviews assessed the benefits of flu vaccine in preventing influenza and ILI in healthy elderly3 and healthy children.4 These Cochrane reviews report a greater benefit from flu vaccine in these two vulnerable population. Among the healthy elderly, 29 people would need to be vaccinated to avoid one case of influenza (ARD: 3.4%; RR: 0.42, 95%CI, 0.27 to 0.66; low‐certainty evidence) and 42 people would need to be vaccinated to prevent one case of ILI (ARD: 2.5%; RR: 0.59, 95%CI, 0.47 to 0.73; moderate‐certainty evidence).3 The impact is even more pronounced in children (2 to 16 years of age) with an NNT of 5 for preventing influenza (ARD: 19%; RR:0.36, 95%CI, 0.28 to 0.48; high‐certainty evidence) and NNT of 12 for preventing ILI (ARD:8%, RR: 0.72, 95%CI, 0.65 to 0.79; moderate‐certainty evidence).4
Caveats
A limitation of the presented data is the heterogenity of the included studies. The degree of benefit in reduction of ILI varied across different settings due to inconsistent definition and symptom classification for ILI.2 In most trials, influenza infection was confirmed by virus isolation from culture, a four-fold antibody increase in the acute or convalescent-phase or in post-vaccination phase.2The vaccine strength varies year by year as the vaccine for one year is generally developed based on the flu strain from the previous year. Therefore, flu vaccine’s efficacy in preventing influenza may vary from one year to another.
Another source of bias in the flu vaccine trials could be originated from using placebo instead of no-intervention as controls; as placebo effect in this situations might reduce the magnitude of benefits.
It must be noted that these meta-analyses assessed the effectiveness of influenza vaccine in healthy individuals only. The vaccine is expected to have a greater protective effect on patients with multiple comorbidities and those who are immunocompromised. In addition, it is hard to factor in the role of herd immunity in a meta-analysis. Herd immunity refers to the protective effective of mass vaccination in prevention of the spread of contagious diseases. If sufficient people are immune to a disease through vaccination, non-immune individuals could indirectly be protected against the disease.5
Conclusion: Because of the evidence of significant benefit of flu vaccine in preventing influenza and influenza-like illness in healthy individuals and absence of serious adverse events, we have assigned a color recommendation of Green (Benefit > Harm) to this vaccine. The efficacy of the flu vaccine in preventing influenza is even more pronounced in healthy children and healthy elderly.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Sally Liang, MDSupervising editor: Shahriar Zehtabchi, MD
Published/Updated
References:
Opiate Analgesia for Acute Abdominal Pain
Benefits in NNT
Significant reduction in hospital length-of-stay
Harms in NNT
No significant adverse events or morbidity
No significant difference in physical exam findings
No significant increase in incorrect diagnosis
No significant difference in treatment decision errors
View As:
Source
Manterola C, Vial M, Moraga J, Astudillo P. Analgesia in patients with acute abdominal pain. Cochrane Database Syst Rev 2011:CD005660.Study Population: 922 adult and adolescents (age > 16 years) with acute abdominal pain from 8 trials
Efficacy Endpoints
Rate of accurate management decisions (primary), change in pain intensity and comfort level (secondary)Harm Endpoints
Physical exam changes, treatment decision errors, incorrect diagnosis, morbidity, hospital length-of-stayNarrative
Acute abdominal pain (AAP) is the most common reason for U.S. emergency department (ED) visits (6-10%),1, 2 and the incidence is rising.3 Admission rates approach 25%, and the majority (44% - 59%) of patients are treated with an opiate/opioid analgesic, with morphine being administered most commonly (20%).4Synthetic opiates exert analgesia effects by stimulating pain-inhibitory neurons and inhibiting pain-transmission neurons through interactions with mu receptors in the central nervous system (CNS).5 It was once thought that blocking the somatic efferent fibers conducting messages to the abdominal muscles and skin may alter crucial peritoneal signs (rigidity, guarding, rebound) that may be associated with intra-abdominal pathology requiring surgical intervention.5 Thus, providing opiate analgesia to patients with AAP was historically discouraged until a surgeon had decided to operate for fear of impeding proper diagnosis. This arose from an era when morphine was administered as grains of morphine tartrate, where 1 grain equated 64mg of morphine sulfate, and was famously promoted by the widely read text “Cope’s Early Diagnosis of the Acute Abdomen” (1921). It also led to the common problem of inadequate pain management (oligoanalgesia) for ED patients with AAP. Only 50-66% ED patients with abdominal pain receive analgesia.3, 4, 6, 7, 8 Average analgesia wait times vary considerably and may be longer than the total mean ED length-of-stay for all comers,2, 3 with one study reporting time to analgesia of 4.1 hours (mild), 1.85 hours (moderate), and 1.37 hours for severe AAP.9
The referenced meta-analysis assesses the impact of opioid analgesic administration to patients with AAP on the diagnostic process.10 It includes 8 randomized clinical trials (RCTs; 922 participants). The studied analgesics were morphine sulfate (5-15 mg; 6 studies), tramadol (1 mg/kg; 1 study), and papaveretum (20 mg; 1 study) as compared to saline control. Papaveretum (Omnopon®, Roche Products Ltd, Welwyn Garden City, UK-England) contains a mixture of opium alkaloid salts including phenanthrene groups (morphine, codeine) that exert actions on CNS mu receptors, and anti-spasmodic isoquinoline groups (papaverine). The formulation used in the included study (prior to 1993) also contained noscapine.11 Assessed outcomes included changes in physical examination findings, pain, adverse events, and diagnostic accuracy as determined by concordance of diagnoses by the emergency physician (EP) and discharging physician. Management decision errors were determined by discordance between the EP’s perceived need for surgical vs. medical management versus the treatment administered during hospitalization.
Pre-treatment pain intensity measured by 10 cm visual analogue scale (VAS) was similar between opioid and placebo groups 5.29 ± 2.32 versus 5.18 ± 2.04 (95% confidence interval [CI] -0.01, 0.26; p=0.058).10 Pain intensity was significantly lower in opioid-treated patients in aggregate (mean difference [95%CI]: -2.00 [-2.89, -1.10]), as well as for each agent individually.10 Additionally, when compared to placebo, no change in physical exam findings (5 trials, 328 participants) was observed individually or in aggregate (RR 1.23, 95% CI, 0.69 to 2.20; quality of evidence: high, or by treatment agent (morphine, tramadol).10 Treatment decision errors (3 studies, 488 participants) did not differ between groups in aggregate (RR 0.77, 95% CI, 0.23 to 2.54; quality of evidence: high), or by individual agent (morphine, papveretum).10 Moreover, the likelihood of an incorrect diagnosis (6 studies, 786 participants) did not differ between groups in aggregate (RR 0.86, 95% CI, 0.57 to 1.29; quality of evidence: high), or by individual agent (morphine, papveretum).10 Patient morbidity (nausea or vomiting; 4 studies, 581 participants) did not differ between groups (RR 5.14, 95% CI, 0.26 to 103.37; quality of evidence high). Lastly, hospital length-of-stay in days (1 study, 100 participants) was shorter in the treatment group (mean difference [95%CI]: -1.00 [ -1.52, -0.48]; quality of evidence: high).
Caveats
Despite evidence that the use of opioid analgesics in patients with AAP improves analgesia without changing physical exam findings or increasing treatment decision errors, morbidity, or likelihood of incorrect diagnosis, there are caveats to consider.10 First is the confusion that may arise from applying the number needed to treat (NNT) concept to large absolute risk reductions. For example, the NNT of 5 in this case could be viewed by some to mean that for every five-people treated with opiates one person benefited (more than control), a statement that may seem incongruent with clinical experience. It is possible that everyone in the intervention group had more pain relief than controls, but in aggregate the mean VAS difference between groups was 20%.Next, significant heterogeneity existed for the variables change in pain intensity (I2 95%) and errors in treatment decisions (I2 55%), which indicate variation pain interpretation between studies, and therefore treatment decision making. The VAS scale is sensitive due to the large number of response categories; however, it may produce unreliable assessments across patient populations owing to variations in scale interpretation. Moreover, the risk-of-bias was high for four studies due to differing research objectives, small sample sizes and inadequate randomization which could invalidate the results.10 Additionally, studies did not report the impact of opioid administration on ED length-of-stay or time to decision for surgery,1 highlighting the need for additional studies. Moreover, studies assessing the impact of other commonly used analgesics (fentanyl, hydromorphone, codeine, hydrocodone, oxycodone) were not identified. Furthermore, the effect of non-intravenous administration routes (intramuscular or oral) remains unknown, as does the impact of pre-existing opiate analgesia taken prior to ED presentation, and the impact of non-opioid analgesic co-administration remains unclear. Lastly, the impact of the common occurrence of mixing different opioid agents in the same patient remains unclear.
A Green color recommendation is based on the evidence of high-quality evidence supporting the benefits that opioid analgesia for patients with AAP improves pain management, without changing physical exam findings or increasing treatment decision errors, patient morbidity, or likelihood of incorrect diagnosis.
Conclusion: Treating patients with acute abdominal pain with opioid analgesics improves analgesia without increasing the risk of diagnosis error or increasing treatment decision errors.
Author
Karissa A. Lambert, MD; Andrew C. Miller, MDPublished/Updated
November 16, 2018References:
Coadministration of Probiotics With Prescribed Antibiotics for Preventing Clostridium difficile Diarrhea
Benefits in NNT
42
1 in 42 for preventing C. difficile–associated diarrhea
12
1 in 12 in high risk patients
Harms in NNT
No significant harm
View As:
Source
Goldenberg JZ, Yap C, Lytvyn L, et al. Probiotics for the prevention of Clostridium difficile-associated diarrhea in adults and children. Cochrane Database Syst Rev 2017;12: CD006095Study Population: 9,955 adults (>18 years) and children (0 to 18 years of age) from 39 trials receiving antibiotic therapy for any reason
Efficacy Endpoints
Clostridium difficile–associated diarrhea, probiotic adverse events, antibiotic-associated diarrhea, length of hospital stay, hospital survival, and 30-day survivalHarm Endpoints
Abdominal cramping, flatulence, soft stools, nausea, fever, and taste disturbanceNarrative
Clostridium difficile colitis is an opportunistic infection that occurs in individuals whose normal gut microbiota has been disrupted. Antibiotics can disturb the normal intestinal microflora, thereby reducing pathogen resistance to the Gram-positive, anaerobic, spore-forming bacillus. When a person takes antibiotics, good germs that protect against infection are destroyed for several months.1 During this time, patients can get sick from C. difficile, a Gram-positive anaerobic spore-forming bacillus picked up from contaminated surfaces or spread from a health care provider’s hands.1 C. difficile infection incidence in the United States has increased dramatically since 2000. In the United States, hospitalizations for C. difficile infection among nonpregnant adults doubled from 2000 through 2010 and were projected to continue to increase in 2011 and 2012.2 On the basis of data from U.S. death certificates, C. difficile infection is the leading cause of gastroenteritis-associated death and was estimated to cause 14,000 deaths in 2007.1, 2 Although almost half of infections occur in people younger than 65, more than 90% of deaths occur in those 65 and older.1 C. difficile has become the most common cause of health care–associated infections in U.S. hospitals, and the excess health care costs related to C. difficile infection are estimated to be as much as $4.8 billion for acute care facilities alone.2 All classes of antibiotics have been associated with C. difficile infections, and elevated risk may continue for up to 3 months postcessation. The spectrum of C. difficile–related disease varies from asymptomatic intestinal colonization, diarrhea, colitis, and pseudomembranous colitis with toxic megacolon.3Probiotics are live organisms (bacteria or yeast) thought to confer a health benefit by improving host microbial balance, counteracting potential disturbances in intestinal flora associated with antibiotic use, and reducing the risk of colonization by pathogenic bacteria.4 They may prevent C. difficile-associated diarrhea (CDAD) through immune system stimulation, impact on the intestinal microbiome, direct inhibition of C. difficile growth, and toxin neutralization.4
The Cochrane meta-analysis cited here assessed the effectiveness of probiotics for decreasing the risk of CDAD among participants taking antibiotics.5 They included 39 randomized clinical trials (RCTs; 9,955 participants).5 A complete case analysis (i.e., participants who completed the study) among trials investigating CDAD (31 trials, 8,672 participants) suggests that probiotics reduce the incidence of CDAD when taken with antibiotics (RR = 0.4; 95% confidence interval [CI] = 0.3–0.52; absolute risk difference [ARD] = 2.4%; NNT = 42; GRADE quality of evidence, moderate). In other words, among patients receiving antibiotics, one would need to treat 42 patients with probiotics to prevent one case of CDAD.
The authors of the Cochrane meta-analysis performed a post hoc subgroup analysis on baseline risk of CDAD (low 0% to 2%; moderate 3% to 5%; high > 5%), determined by the event risk in placebo or no treatment group.5 Among studies with a baseline risk > 5%, the benefit of probiotics was even more pronounced (13 trials, 2,454 participants; RR = 0.30; 95% CI = 0.21–0.42; ARD = 8.1%; NNT = 12; GRADE quality of evidence, moderate). This means that one would need to treat 12 patients at high risk for CDAD with probiotics to prevent one case of CDAD resulting from antibiotic use.
In the Cochrane meta-analysis, 15 studies (n = 1,214) reported on the secondary endpoint of detection of C. difficile independent of presence of CDAD.5 Of these, 13 were placebo controlled and two trials used a no treatment control arm. The overall pooled results using a complete case approach did not show a statistically significant reduction in detection of C. difficile in the stool. C. difficile was detected in 15.5% (98/633) of the probiotics group compared to 17.0% (99/581) of the placebo or no treatment control group (RR = 0.86; 95% CI = 0.67–1.10; random effects).5 Eleven of the 15 studies were rated as having a high or unclear risk of bias. No statistically significant heterogeneity was detected for this comparison (p = 0.88; I2 = 0%).5
Caveats
This systematic review and meta-analysis assesses the safety and efficacy of probiotics for decreasing the risk of CDAD in adults and children taking antibiotics.5 Unfortunately, the analysis does not control for type or strain of the probiotics or for antibiotic class or administration route. In addition to the 39 trials (9,955 participants) identified by the Cochrane group, we identified three small relevant randomized clinical trials that were published after completion of the Cochrane review.6, 7, 8 These trials each studied a specific type of probiotic, exclusively in admitted patients, but did not show any significant difference for risk of CDAD between groups. They did not control for baseline risk and likely suffered from Type II error.6, 7, 8Unfortunately, the meta-analysis does not provide a clear answer regarding the safety of probiotics. Adverse events (AEs) were a composite endpoint including pooled data on abdominal cramping, flatulence, soft stools, nausea, fever, and taste disturbance. The data for AEs are reported to be of low quality by the Cochrane review. Although significantly fewer AEs were reported in the probiotic compared to the control group (14.3% vs. 17.0%; RR = 0.83; 95% CI = 0.71–0.97), moderate heterogeneity was detected (p = 0.005; I2 = 49%).5 Additionally, 18 (56%) studies were rated as having a high or unclear risk of bias.
Conclusions are limited by the degree of missing data in the included trials. Moreover, the meta-analysis controlled for neither probiotic nor antibiotic types. In this manner one assumes that all probiotic treatments and antibiotic exposures are equivalent in their effect and that any variation in effect is due to chance.5 However, it is unknown if all probiotics are equally efficacious, if specific dosing thresholds need be met, or if effective probiotic dose varies based on antibiotic class being administered. Indeed, a meta-analysis by Allen et al.9 found that a formulation of Lactobacillus acidophilus + Lactobacillus casei + Lactobacillus rhamnosus showed positive results, whereas Saccharomyces boulardii alone and a combination of Lactobacillus acidophilus + Bifidobacterium bifidum + Bifidobacterium lactis was shown not to be effective.9
Finally, the Cochrane review included a mix of inpatients and outpatient subjects.5 However, the subgroup analysis did not show any significant impact for hospitalization. Regardless of this finding, it is plausible that patients admitted to the hospitals or intensive care units might respond differently to probiotics.
The green color recommendation is based on evidence of moderate quality data supporting benefits from probiotics in preventing CDAD in patients on antibiotics, in the absence of evidence of serious harms associated with their use.
Conclusion: Based on this systematic review and meta-analysis of 31 randomized controlled trials including 8,672 patients, moderate certainty evidence suggests that probiotics are effective for preventing CDAD. Although they did not describe how risk was determined, post hoc subgroup analyses indicated that probiotics are effective among trials with a CDAD baseline risk > 5% (moderate certainty evidence), but not among trials with a baseline risk ≤ 5% (low to moderate certainty evidence). No significant harm was reported (low-quality evidence). The effect of probiotic type, dose, antibiotic, and antibiotic route remains unclear.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
See theNNT.com's previous reviews of this topic:
Probiotics for Preventing C. Difficile Diarrhea, February 11, 2013
Author
Ahmed Hamed, MD; Andrew C. Miller, MDSupervising Editor: Shahriar Zehtabchi, MD.
Published/Updated
November 5, 2018References:
Hormone Therapy for Primary Prevention of Cardiovascular Disease in Postmenopausal Women
Benefits in NNT
No benefit observed
Harms in NNT
165
1 in 165 had a stroke
118
1 in 118 had a blood clot in the leg or lung
242
1 in 242 had a blood clot in the lung
View As:
Source
Schierbeck LL, Rejnmark L, Tofteng CL, et al. Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: randomised trial. BMJ. 2012; 345: e6409.Study Population: 40,410 postmenopausal women (healthy or with preexisting cardiovascular disease) across 19 randomized controlled trials that examined the use of oral hormone therapy (estrogen with or without progesterone taken for at least six months) compared with placebo
Efficacy Endpoints
Death from any cause; death caused by heart attack; nonfatal heart attackHarm Endpoints
Stroke, thromboembolic event (deep venous thrombosis plus pulmonary embolism) or pulmonary embolismNarrative
In 2012, the Danish Osteoporosis Prevention Study published a study in the British Medical Journal that compared hormone therapy with placebo and found a decreased risk of heart failure, heart attack, and death from any cause.1 They also found no increased risk of cancer, blood clots, or stroke. Although the Danish cohort studied younger women and used different preparations of hormone therapy, the results were in direct contrast to most of the studies analyzed in a 2005 Cochrane review. This 2005 review found no benefit of hormone therapy in preventing cardiovascular disease, but noted significant increases in the risk of developing stroke and blood clots in a leg or lung.2The 2015 Cochrane review added six additional studies (compared with the initial review published in 20133), for a total of 19 randomized controlled trials, consisting of 40,410 postmenopausal women.4 The studies examined the use of oral hormone therapy (estrogen with or without progesterone) taken for at least six months, compared with placebo, to determine the effect on death from any cause, and death caused by heart attack, nonfatal heart attack, angina, revascularization, stroke, and blood clot in a leg or lung. The current review found no benefits in the primary prevention of heart attack (fatal or nonfatal), angina, revascularization, or death due to any cause. However, they did find increases in the risk of stroke (relative risk [RR] = 1.32; 95% confidence interval [CI], 1.12 to 1.56; absolute risk increase [ARI] = 0.6%; number needed to harm [NNH] = 165), blood clot in a leg or lung (RR = 1.92; 95% CI, 1.24 to 2.99; ARI = 0.8%; NNH = 118), and blood clot in a lung (RR = 1.89; 95% CI, 1.17 to 3.04; ARI = 0.4%; NNH = 242) in patients receiving hormone therapy for at least six months.
Caveats
In response to the Danish study,1 the authors of the Cochrane review performed a subgroup analysis to assess for the possibility that women less than 10 years postmenopause may have a different response than women more than 10 years postmenopause. The current Cochrane analysis revealed that although older women (more than 10 years postmenopause) still have increased risk of stroke (RR = 1.21; 95% CI, 1.06 to 1.38; ARI = 1%; NNH = 102) and blood clot in a leg or lung (RR = 1.96; 95% CI, 1.37 to 2.80; ARI = 1%; NNH = 101), younger women (less than 10 years postmenopause) had some benefit, including lower rates of death (RR = 0.70; 95% CI, 0.52 to 0.95; absolute risk reduction [ARR] = 0.7%; number needed to treat [NNT] = 146) and lower rates of fatal and nonfatal heart attacks (RR = 0.52; 95% CI, 0.29 to 0.96; ARR = 0.7%; NNT = 133). Yet, younger women still had increased risk of blood clot in a leg or lung (RR = 1.74; 95% CI, 1.11 to 2.73; ARI = 0.5%; NNH = 214). This may explain the contradictory results seen in the Danish study, but the findings of this subgroup analysis were derived from only a few studies that involved many fewer patients than the overall review. Consequently, further studies will be required to determine whether there may be some cardiovascular benefit of hormone therapy in younger postmenopausal women.There are some biases that might have affected the results of the meta-analysis. First, most patients enrolled in the trials were on average older than 60 years at baseline. Given the possibility of time-from-menopause being a possible confounder, this issue could subject the analysis to significant bias. Second, different hormone therapy regimens were used in the trials, introducing heterogeneity in the meta-analysis. The overall quality of evidence, however, is rated as high by the authors of the Cochrane review.4
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Theodore J. Segarra, MD; Michael Ritchie, MD; and Shahriar Zehtabchi, MDPublished/Updated
October 5, 2018References:
Endovascular thrombectomy for ischemic stroke beyond 6 hours from onset of symptoms
Benefits in NNT
3-4
3-4 for functional independence at 90 days
Harms in NNT
35
35 for symptomatic intracranial hemorrhage
View As:
Source
Nogueira RG, Jadhav AP, Haussen DC, Bonafe A, Budzik RF, Bhuva P, et al; DAWN Trial Investigators. Thrombectomy 6 to 24 Hours after Stroke with a Mismatch between Deficit and Infarct. N Engl J Med. 2018;378:11-21.Study Population: 388 patients with large vessel (internal carotid artery or proximal middle cerebral artery) occlusion, mismatch between infarct size and either clinical deficit or ischemic penumbra, and presentation more than 6 hours (up to 16 or 24 hours) after onset of stroke symptoms
Efficacy Endpoints
Disability score at day 90; functional independence (defined as a score of 0-2 on the Modified Rankin Scale at day 90)Harm Endpoints
Death (90-day mortality); symptomatic intracranial hemorrhage; serious adverse eventsNarrative
In ischemic stroke, there are two types of affected brain tissue. The center of the lesion is composed of dead or non-salvageable brain tissue. This core is surrounded by a region of ischemic tissue called the penumbra. The penumbra is the area that theoretically might be salvaged with reperfusion therapy.1,2Endovascular thrombectomy has been shown to reverse the ischemia in this penumbra in certain stroke patients with large vessel occlusion if it is performed within 6 hours of onset of symptoms. However, as the time from symptom onset increases, the chance of an improved neurologic outcome declines.3 The time window for saving ischemic tissue may be different in different patients, suggesting that time may not be the only factor determines the likelihood of benefit from reperfusion. Some investigators believe that in a carefully selected group of patients a substantial amount of brain tissue remains salvageable beyond the 6-hour treatment window.
Recently, two randomized trials1,2 suggested that removing a large vessel clot can improve outcomes even 16-24 hours after the onset of symptoms. One study targeted patients with a clinical deficit that was disproportionately severe relative to the imaged infarct volume, which suggested that symptoms were due to a large area of hypoperfused but not yet infarcted tissue. The other study targeted patients with a large neuroimaging mismatch between ischemic tissue and infarcted tissue. These trials used CT perfusion imaging or MRI diffusion and perfusion scanning paired with automated image post-processing systems to map the affected area of the brain. Both trials were terminated early due to efficacy.
DEFUSE 31 was a multicenter, randomized, open-label trial that was conducted at 38 centers in the United States (Table 1). In this trial, thrombectomy plus medical therapy, as compared with medical therapy alone, was associated with a favorable shift in the distribution of functional outcomes on the Modified Rankin Scale (mRS) at 90 days (odds ratio: 2.77; 95%CI, 1.63-4.70, P<0.001), as well as a higher percentage of patients who were functionally independent, defined as a mRS score of 0-2 (45% vs. 17%, P<0.001, Absolute Risk Difference [ARD]: 28%, NNT:4).
The 90-day mortality rate was 14% in the thrombectomy group and 26% in the medical-therapy group (ARD:12%, Odds ratio:0.55; 95%CI, 0.30–1.02, p=0.05), and there was no statistically significant difference in the frequency of symptomatic intracranial hemorrhage (7% and 4%, respectively; odd ratio:1.47, 95%CI, 0.40–6.55; P=0.75) or of serious adverse events (43% and 53%, respectively; P=0.18) between the groups. Thrombectomy-related complications occurred in two patients (one vessel perforation resulting in subarachnoid hemorrhage and one device-related vasospasm).
DAWN2 was a multicenter, randomized, open-label trial with a Bayesian adaptive–enrichment design and with blinded assessment of end points conducted at 26 centers in the United States, Canada, Europe, and Australia (Table 1). This trial was sponsored by Stryker Neurovascular, which provided funding, thrombectomy devices, performed regulatory monitoring at each site, and provided central database maintenance.
The mean score on the Utility-Weighted Modified Rankin Scale (which weights each category according to patient perception of health-related quality of life) at 90 days was 5.5 in the thrombectomy group as compared with 3.4 in the control group (adjusted difference [Bayesian analysis], 2.0 points; 95% CI, 1.1-3.0). The rate of functional independence at 90 days was 49% in the thrombectomy group, as compared with 13% in the control group (ARD:33%; 95% CI, 24-44; NNT:3). The rate of symptomatic intracranial hemorrhage did not differ significantly between the two groups (6% in the thrombectomy and 3% in the control group, P=0.50), nor did 90-day mortality (19% and 18%, respectively; P=1.00).
The rates of safety end points and serious adverse events — including stroke-related death at 90 days, death from any cause at 90 days, and symptomatic intracerebral hemorrhage — did not differ significantly between the two treatment groups. The rate of neurologic deterioration was lower in the thrombectomy group than in the control group (14% vs. 26%; absolute difference, −12 percentage points; 95% CI, −23 to −1; P = 0.04). Seven patients in the thrombectomy group (7%) experienced procedure-related complications (intramural arterial dissection, arterial perforation, and access-site complications leading to intervention).
The results of these two trials indicate that in patients with good pre-stroke baseline functional status who had large vessel occlusion and a mismatch between infarct size and either clinical deficit or ischemic penumbra and an onset more than 6 hours (up to 16 or 24 hours) before presentation, neurologic outcomes at 90 days could be better with thrombectomy. While time is still the most important element in treatment success, these trials highlight the importance of adding tissue-based criteria in selecting patients for revascularization. The most important harm associated with this strategy is the increased risk of symptomatic intracranial hemorrhage. The combined number of patients with symptomatic intracranial hemorrhage in both trials was 19/306 (6.2%) in intervention groups vs. 10/296 (3.4%) in control groups, ARD: 2.8% (95% CI, 0.7-6.0%), resulting in a number-needed-to-harm (NNH) of 35 (95% CI, 15-149).
Caveats
DAWN and DEFUSE 3 are important trials with significant clinical implications for patients, physicians, and hospitals. These trials employed a new approach to identify potentially salvageable brain tissue (penumbra) and to select stroke patients for revascularization.These findings have potentially serious service implications for stroke systems of care. At the prehospital level, EMS providers would be required to screen suspected stroke patients for possible large vessel occlusion up to 24 hours from last-seen-well (LSW) time, and preferentially transport those meeting eligibility criteria to a center capable of rapid advanced imaging and endovascular therapy.
At the hospital level, stroke centers would need to perform CT perfusion or MRI rapidly in patients up to 24 hours after the onset of symptoms. This might require not just increased equipment, but also advanced software, increased staffing by CT and MRI technologists, and enhanced neuroradiology availability to rapidly interpret these images. It is not clear how many hospitals or even primary stroke centers could feasibly offer these services. In addition, hospitals without endovascular teams would need to put in place systems to rapidly evaluate and transport patients to endovascular-capable hospitals, as necessary.
Such a change in current clinical practice has been estimated to potentially increase the number of patients eligible for thrombectomy by only 2-5%. However, many patients would need to be screened for eligibility (including with advanced imaging). At many hospitals, the costs of maintaining the necessary 24/7 advanced imaging might not be feasible. The cost effectiveness of this approach, as well as scalability outside of academic medical centers, is thus not yet clear. In addition, recent publication of the use of intravenous thrombolysis in patients selected by similar criteria raises the concern for even more complex decision-making in the acute phase.4
It must also be emphasized that the criteria for enrollment in DAWN and DEFUSE 3 were relatively strict and were limited to patients with good baseline functional status, large vessel ischemic strokes limited to proximal carotid artery or middle cerebral artery, and large enough mismatches between the size of ischemic vs infarcted tissue. The enrolled patients had relatively small core infarctions. It is likely that most stroke patients would not meet these criteria.
A major limitation in generalizing the findings of these trials is the method of determining infarct size. These trials employed sophisticated software (RAPID, iSchemaView) with DWI or CTP imaging. It is not clear that centers without this software, or rapid MRI/CTP, could achieve similar results. The more widely available method of determination of perfusion mismatch (volumetric or qualitative) should be evaluated in identifying similar patients and producing similar outcomes.
While we have assigned a GREEN color recommendation, we caution the readers about patient selection. The inclusion criteria for this treatment do not apply to majority of stroke patients. In addition, the complexity of the technology and resource demands may limit which centers can safely and effectively employ this approach.
Author
Shahriar Zehtabchi, MD; Joshua N. Goldstein, MD, PhDPublished/Updated
September 25, 2018References:
Epinephrine for Out-of-Hospital Cardiac Arrest
Benefits in NNT
7
1 in 7 achieved ROSC prior to hospital arrival
No benefit for survival to hospital discharge or survival at one month
Harms in NNT
83
1 in 83 had a worse long-term neurological outcome
View As:
Source
Loomba RS, Nijhawan K, Aggarwal S, Arora RR. Increased return of spontaneous circulation at the expense of neurologic outcomes: Is prehospital epinephrine for out-of-hospital cardiac arrest really worth it? Journal of Critical Care. 2015;30:1376-1381.Study Population: Total of 655,853 patients from 13 observational studies and one randomized, controlled trial (RCT) involving patients who experienced out-of-hospital cardiac arrest.
Efficacy Endpoints
Pre-hospital ROSC, survival to hospital discharge, survival at one monthHarm Endpoints
Long-term neurological outcome, defined as Cerebral Performance Category (CPC) score of 1-2 (corresponding to independence in Activities of Daily Living)Narrative
The original data supporting the use of epinephrine for cardiac arrest is rooted in a poorly controlled canine study1 from the 1960s. The Advanced Cardiac Life Support (ACLS) recommendation that it “may be reasonable” to use epinephrine in cardiac arrest continues in contemporary practice. Yet, when the ACLS guidelines2 are read carefully, they state that “for both survival to discharge and survival to discharge with good neurological outcome, there was no benefit” to receiving epinephrine.Physiologically, epinephrine is theorized to promote peripheral vasoconstriction, thereby increasing diastolic pressure and coronary perfusion. Epinephrine also increases myocardial work and metabolic demand and may worsen tachydysrhythmia. Despite the purported physiologic benefits, it appears that epinephrine use for out-of-hospital cardiac arrest (OHCA) increases the rate of ROSC but does not increase the chance of survival (Table). Epinephrine may even worsen the neurological outcome in patients who do survive. Epinephrine use for OHCA may therefore extend suffering and increase end-of-life healthcare costs due to intensive care and prolonged hospitalization without clear patient-centered, long-term benefits. Patients who survive to hospital discharge may be more likely to be dependent on others for care. This presents an obvious ethical quandary, as patient and family preferences may differ greatly with regards to life-prolonging therapies.
Caveats
The source meta-analysis3 incorporated 13 observational studies and only one RCT:4 Jacob et al4 studied 601 patients and showed that epinephrine for OHCA was effective at increasing rate of ROSC (OR 3.4, 95% CI 2.0 to 5.6) and survival to hospital admission (OR 2.3, 95% CI 1.4 to 3.6) but lacked a statistically significant effect on survival to hospital discharge (OR 2.2; 95% CI 0.7 to 6.3). The observational trials included in the meta-analysis3 attempted to control for potential confounders (i.e. time to CPR) by propensity-matching individual study subjects, although bias can never be fully accounted for in any observational study. Similarly, the use of random effects methods cannot fully control for heterogeneity when reporting pooled effects in meta-analyses, and 3/4 of reported outcomes had exceedingly high heterogeneity (I2 = 96%). The source of heterogeneity may be patient characteristics, co-interventions, or trial-level, and random effects methods only attempt to adjust for between-trial variability, which can unintentionally inflate the effect of small studies on the pooled results.5 Propensity-matched outcomes in individual studies were generally in agreement with the pooled outcomes in this meta-analysis3 with the exception of survival at one month. The pooled results for one month survival may be confounded by the negative effect of smaller studies that were in favor of withholding epinephrine.Specific effect modifiers in the included studies such as the timing and dosing of epinephrine administration may have influenced treatment effects on resuscitation outcomes. Earlier epinephrine administration has been associated with more favorable outcomes.6, 7, 8
The ‘standard’ one mg of epinephrine given in cardiac arrest is not weight-based and therefore can have a differential physiologic effect depending on the individual patient.9 The meta-analysis3 included some trials that used ‘high dose’ (0.1 to 0.2 mg/kg) epinephrine, which may have contributed negatively to pooled outcome results. Epinephrine administered at higher doses may be harmful10 and is not recommended by current guidelines.2
Loomba et al3 defines positive neurologic outcome as a cerebral performance category (CPC) of 1 or 2, which represents mild or moderate cerebral dysfunction but ability to independently perform activities of daily living. This is a validated method of measuring neurological outcome; however, trials included in the systematic review3 measured the CPC at different time points (hospital discharge, one month, etc.), resulting in significant heterogeneity in the data. The “recovery time” prior to CPC assessment is an important confounder when determining neurological outcome as are post-ROSC interventions such as targeted temperature management and urgent cardiac catheterization, none of which were adjusted for in the individual trials of this meta-analysis.3 Therefore, the results of the meta-analysis pertaining to this outcome should be interpreted with caution. The data discussed in Loomba et al3 are primarily applicable to cardiac arrests that occur out-of-hospital, and conclusions may not apply to patients that experience cardiac arrest in the setting of an emergency department, hospital floor, intensive care unit, or operating room. In these more controlled hospital settings, epinephrine is more likely to be given earlier along with prompt defibrillation and high-quality CPR, and therefore its use for in-hospital cardiac arrests may result in different outcomes. Finally, this systematic review3 analyzed only adult patients, and its conclusions should not be applied to the pediatric population.
Although in the past, there may have been barriers to performing true RCTs that must withhold potentially life-saving, guideline-recommended, “standard of care” from a control group, a double-blind, RCT (PARAMEDIC-2)11 was recently published and largely agrees with the meta-analysis.3 After enrolling 8,014 patients and following them in pragmatic fashion, PARAMEDIC-2 demonstrated that epinephrine increases survival to hospital admission and 30 days, with an NNT of 112 to prevent one death at 30 days. However, the additional proportion that received epinephrine and survived did so with severe neurological disability, defined as 4 or 5 on the modified Rankin scale. There was no evidence of benefit in the proportion of patients who survived to hospital discharge with a favorable neurologic outcome (OR 1.18, 95% CI 0.86 to 1.61), and the lack of effect persisted at 3 months. This confirms that the use of epinephrine for OHCA is not patient-centered (as defined by patients and the public in preparation for the PARAMEDIC-2 trial) and will cause prolonged suffering in a severely disabled state.
In summary, we chose a color recommendation of “Red” for epinephrine administration in OHCA. There is no patient-centered benefit and probable harm due to increased survival with worse long-term neurological function.
The original manuscript was published in Academic Emergency Medicine as part of the partnership between TheNNT.com and AEM.
Author
Kyle Kelson, MD and Ian S. deSouza, MDSupervising editors: Kabir Yadav, MD; Shahriar Zehtabchi, MD
Published/Updated
August 13, 2018References:
Lidocaine for Reducing Pain on Injection of IV Propofol
Benefits in NNT
3
1 in 3 no longer experience pain on propofol injection
Harms in NNT
No significant harm
View As:
Source
Euasobhon P, Dej-arkom S, Siriussawakul A, Muangman S, Sriraj W, Pattanittum P, Lumbiganon P. Lidocaine for reducing Propofol-induced pain on induction of anaesthesia in adults. Cochrane Database of Systematic Reviews 2016, Issue 2. Art. No.: CD007874.Study Population: 10,350 participants from 82 studies were included for quantitative analysis, (age range 13 to 89 years) with American Society of Anesthesiologists (ASA) status I-III undergoing elective surgery who received IV propofol for induction of anesthesia.
Efficacy Endpoints
Overall pain & high intensity pain (according to the definition of the scoring system of each included trial)Harm Endpoints
Thrombophlebitis, cardiac arrhythmia, allergic reaction, or local anesthetic toxicityNarrative
Localized pain at the injection site and up the course of the cannulated vein upon IV propofol injection is an unpleasant side effect that is frequently seen during induction of anaesthesia and during moderate sedation used in Emergency Departments across the globe. IV lidocaine injection has been commonly used to attenuate this effect. Many studies have reported that lidocaine is effective in reducing the incidence and severity of pain. This topic was recently analyzed by the Euasobhon et al1 in the 2016 Cochrane Collaboration systematic review and is the primary source for this summary.A total of 85 studies were included in this systematic review, 82 of which (10,350 participants) were eligible for quantitative analysis. All studies were single-center and were performed in a range countries worldwide. 12/84 trials used co-induction medications as an adjunct in both groups: remifentanil (4 trials), nitroglycerine ointment (1 trial), 50% Nitrous Oxide (3 trials), sevoflurane (1 trial), dexamethasone (1 trial), ketamine (1 trial) or dexmedetomidine (1 trial). 82 of the studies were assessed to be of satisfactory methodological quality to be included in the meta-analysis. All compared propofol to placebo (saline) and participants were randomly allocated to placebo or treatment groups.
Administration of lidocaine, either mixed with propofol or given IV before propofol, significantly reduced the overall incidence of pain associated with propofol injection when compared to placebo: 30% vs. 64%; absolute risk difference (ARD): 34%; NNT: 3. Similarly, lidocaine reduced the risk of experiencing high-intensity pain during propofol injection: 12% vs. 38%; ARD: 26%; NNT: 4. ‘High-intensity pain’ included moderate pain, severe pain, pain mentioned when questioned and accompanied by behavioral signs, or pain reported spontaneously, not as a result of questioning, pain associated with grimacing, withdrawal movement of forearm or tears. Regarding lidocaine/propofol admixture, subgroup analyses using different doses of lidocaine admixed with the propofol i.e. ≤20 mg or ≤0.2 mg/kg, or >20 mg or >0.2 mg/kg, found no statistically significant difference between the two – both reduced pain.
Regarding lidocaine pretreatment with IV injection prior to propofol, during subgroup analyses they compared four different techniques of lidocaine pre-treatment administration: 1. Low dose lidocaine pretreatment alone (≤ 20 mg or ≤ 0.2 mg/kg), 2. High dose lidocaine pretreatment alone (> 20 mg or > 0.2 mg/kg), 3. Low dose lidocaine pretreatment with venous occlusion upstream (≤ 20 mg or ≤ 0.2 mg/kg), 4. High dose lidocaine pretreatment with venous occlusion upstream (> 20 mg or > 0.2 mg/kg). All of these techniques decreased pain on injection. However low dose lidocaine pretreatment without venous occlusion appeared to be the least effective of the four “pretreatment” techniques (OR 0.40, 95% CI 0.29 to 0.55, seven studies, 713 participants, high-quality evidence). Whilst high dose lidocaine pretreatment with venous occlusion was the most effective of the pretreatment techniques (OR 0.11, 95% CI 0.09 to 0.15)
Thirty two of the studies commented on incidence of adverse events. Thrombophlebitis was the only reported adverse effect and was noted in only two studies, the incidence of which was not significantly different between lidocaine and placebo groups.
Caveats
The systematic review reports the quality of evidence to be high, with a low risk of bias. The only area with a low rating for quality of evidence was the incidence of adverse events. It is not possible to estimate accurately the harms and adverse events associated with this practice, as only a few trials included in the systematic review assessed adverse events and they suffered from serious imprecision and inconsistency.The trials analyzed in the systematic review reported here included trials that studied propofol as an induction agent. Propofol is frequently used for procedural sedation in the emergency department (ED). The safety and efficacy of lidocaine to reduce pain associated with propofol injection could be similar during moderate sedation. A literature search revealed no papers related to lidocaine use to attenuate propofol-related injection pain in settings outside of the operating theatre. However, ED-based trials should confirm these findings before they are generalized to ED procedural sedation.
Author
Samuel Desbruslais, MBBSPublished/Updated
References:
Nebulized Hypertonic Saline for Bronchiolitis
Benefits in NNT
5
1 in 5 avoided hospitalization
Harms in NNT
None
View As:
Source
Zhang L, Mendoza-Sassi RA, Klassen TP, Wainwright C. Nebulized hypertonic saline for acute bronchiolitis: a systematic review [published correction appears in Pediatrics. 2016;137(4):pii:e20160017]. Pediatrics. 2015;136(4): 687-701.Study Population: Children younger than 24 months with bronchiolitis (3,209 participants in 24 trials). Most trials excluded patients who required mechanical ventilation, intensive care, or those who had oxygen saturation less than 85% on room air.
Efficacy Endpoints
Length of hospital stay, rate of hospitalizationHarm Endpoints
Tachycardia, hypertension, pallor, tremor, nausea, vomiting, diarrhea, and acute urinary retentionNarrative
Bronchiolitis is the most common lower respiratory tract infection in infants, with respiratory syncytial virus being the leading cause. Airway edema and mucus plugging are believed to be the pathologic processes causing morbidity in cases of viral bronchiolitis. Supportive treatment is the standard of care. In addition, nebulized hypertonic saline may be beneficial in relieving symptoms.A systematic review included double-blind, randomized, controlled clinical trials evaluating the effect of nebulized hypertonic (3% or higher) saline solution alone or in conjunction with bronchodilators in infants with acute bronchiolitis compared with nebulized normal (0.9%) saline.1 Nebulized hypertonic saline resulted in a statistically significant reduction in length of hospital stay (mean difference: –0.45 day; 95% confidence interval [CI], –0.82 to –0.08). Nebulized hypertonic saline also reduced the risk of hospitalization by 20% compared with 0.9% saline (relative risk [RR] = 0.80; 95% CI, 0.67 to 0.96). No significant adverse events related to hypertonic saline inhalation were reported.
The lead author of this systematic review published a Cochrane review on the same topic in 2013.2 That meta-analysis showed a mean reduction of 1.2 days (95% CI, 0.8 to 1.5 days) in the length of hospital stay and no significant difference in the rate of hospitalization.2 We chose to write our summary based on the 2015 meta-analysis because it is more recent and includes several recent trials and approximately 2,000 more patients than the 2013 Cochrane review.
A 2014 meta-analysis reported an approximately one-day decrease in the length of hospital stay (weighted mean difference = –0.96; 95% CI, –1.38 to –0.54) in patients who received nebulized hypertonic saline compared with normal saline.3 This meta-analysis also showed a significant decrease in hospital admission rate (RR = 0.59; 95% CI, 0.37 to 0.93) after receiving nebulized hypertonic saline.
A 2017 randomized controlled trial enrolling 777 patients with bronchiolitis failed to show any significant difference in rate of hospital admission or length of hospital stay between the groups (nebulized hypertonic saline and nebulized normal saline).4 No significant adverse events related to hypertonic saline inhalation were observed in the trials reported in systematic reviews. No patients withdrew because of adverse events or clinical deterioration.
Caveats
The results of three large meta-analyses of randomized double-blind clinical trials suggest a high quality of evidence and show some benefits in using nebulized hypertonic saline compared with normal saline in children with bronchiolitis.1,2,3 However, the most concerning issue arising from reviewing the data is that most of the trials published after 2013, including two large multicenter European trails, have reported negative results.4,5,6,7,8, Heterogeneity among the trials and existence of effect modifiers could be responsible for this inconsistency.The authors of the 2015 review offer an explanation for this discrepancy among the trials.1 A subgroup analysis performed by these authors found that trials in which virologic testing was available showed a significantly greater effect size of nebulized hypertonic saline (measured by reduction of length of hospital stay and rate of hospitalization) than trials without such testing. Thus, the diagnostic accuracy of bronchiolitis may affect the treatment outcomes.
The only consistent finding among all trials is the absence of any serious adverse event associated with the use of hypertonic saline. Considering the prevalence of bronchiolitis and the financial and emotional cost of hospitalization, possible reduction in admission rate and hospital stay with minimal adverse events provides enough evidence to recommend this treatment for acute bronchiolitis.
Most trials excluded patients who required mechanical ventilation or intensive care, and those who had oxygen saturation less than 85% on room air. Therefore, the findings of this review might not apply to infants with more severe bronchiolitis.
Despite reported benefits associated with the use of hypertonic saline for bronchiolitis, we have chosen a yellow recommendation because the more recent trials and systematic reviews have shown either much smaller effect size or no benefit at all compared with the older trials.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
See theNNT.com's previous reviews of this topic:
Nebulized Hypertonic Saline for Bronchiolitis in Infants, March 10, 2011
Author
Gholamreza Roodsari, MD; Shahriar Zehtabchi, MDPublished/Updated
July 10, 2018References:
Antibiotics for Otitis Media in Children
Benefits in NNT
0 had less pain at 24 hours
7-20
1 in 7-20 experience decreased pain after 24 hours (based on assessment days 2 to 12)
33
0 avoided recurrence of infection or hearing loss at 3 months
11
1 in 33 avoided tympanic membrane perforation
1 in 11 avoided contralateral ear infection
0 avoided serious complications (e.g., mastoiditis or meningitis)
Harms in NNT
14
1 in 14 experienced vomiting, diarrhea, or rash
View As:
Source
Venekamp RP, Sanders SL, Glasziou PP, Del Mar CB, Rovers MM. Antibiotics for acute otitis media in children. Cochrane Database Syst Rev. 2015;(6):CD000219.Study Population: Children between two months and 15 years of age from high-income countries enrolled in 13 randomized controlled trials (3,401 participants)1
Efficacy Endpoints
Pain at various time points (24 hours, two to three days, four to seven days, 10 to 12 days), tympanic membrane (eardrum) perforation, contralateral ear infection (in unilateral ear infections), recurrence of ear infection, hearing loss (decreased hearing) after three monthsHarm Endpoints
Medication adverse effects such as vomiting, diarrhea, or rash; serious consequences of ear infection such as mastoiditis or meningitisNarrative
Acute otitis media is a disease that most commonly affects young infants and children. The inflammation and edema caused by the infection can manifest as ear pain or ear fullness, typically accompanied by fever, irritability, and decreased activity and feeding. Complications include tympanic membrane perforation with otorrhea, hearing loss, and recurrent otitis media. More severe complications include mastoiditis, cranial nerve palsies, and meningitis.Currently, there are no universally accepted guidelines pertaining to the use of antibiotics in children diagnosed with otitis media. The guidelines published by the American Academy of Pediatrics recommend an antibiotic prescription for children six months and older with severe signs and symptoms of acute otitis media (moderate to severe otalgia, otalgia for 48 hours or more, or temperature of 102.2°F [39°C] or higher).2 For nonsevere unilateral otitis media, the same guidelines recommend antibiotic treatment or close follow-up based on joint decision-making with parents or caregivers. However, treatment patterns for otitis media differ among physicians. Some prescribe antibiotics liberally, whereas others take a more conservative approach by observing patients for worsening symptoms or development of complications, at which point antibiotics are administered. A Cochrane review compared the effectiveness of antibiotics with expectant observation (no treatment) or placebo. The review analyzes the outcomes of antibiotics vs. placebo and antibiotics vs. expectant observation separately.1
The Cochrane review concludes that antibiotics have no early effect on pain in the first 24 hours (relative risk [RR] = 0.89; 95% confidence interval [CI], 0.78 to 1.01), some effect on pain in the days following (one in seven to 20; RR = 0.70; 95% CI, 0.57 to 0.86 for pain reduction after two to three days; RR = 0.76; 95% CI, 0.63 to 0.91 for pain reduction after four to seven days; RR = 0.33; 95% CI, 0.17 to 0.66 for pain reduction after 10 to 12 days compared with placebo), and some beneficial effect on the number of children with tympanic membrane perforation (one in 33; RR = 0.37; 95% CI, 0.18 to 0.76) or episodes of contralateral ear infection (one in 11; RR = 0.49; 95% CI, 0.25 to 0.95), compared with placebo.1
A meta-analysis using patient data from six high-quality randomized controlled trials is also included in the Cochrane review.1 This meta-analysis concludes that the risk of a prolonged course of antibiotics was twice as high for children younger than two years with bilateral acute otitis media than for children two years and older with unilateral acute otitis media, indicating that antibiotics might be beneficial in resolution of symptoms in this particular subgroup.3
Caveats
The most important caveat is that all trials included in the Cochrane review were conducted in high-income countries. Thus, the results might not be generalizable to lower-income countries. In populations with a higher risk of mastoiditis or with lower immunization coverage, antibiotics might prevent serious complications.4Another notable caveat is the inclusion of patients who received delayed antibiotics in the expectant observation group. In some trials, patients assigned to the observation group were given a prescription but were instructed to withhold antibiotics for 72 hours and to initiate treatment only if symptoms persisted. Delayed antibiotic use might have affected the benefit or harm outcomes in such trials.
Not all trials had specific protocols for pain management (e.g., ibuprofen, acetaminophen). Given that pain reduction was the primary outcome in most trials, the presence or absence of a pain control protocol or of clear instructions for the parents could have influenced the outcomes.
A review of the literature indicates that physicians overdiagnose otitis media, which may result in underestimation of a treatment effect.4 A rational clinical examination sys- tematic review showed that on pneumatic otoscopy, cloudy (adjusted likelihood ratio [LR] = 34; 95% CI, 28 to 42), bulging (adjusted LR = 51; 95% CI, 36 to 73), and distinctly immobile (adjusted LR = 31; 95% CI, 26 to 37) tympanic membranes were the most useful signs of otitis media.5
The methodologic quality of the studies included in the review was deemed to be high, and the evidence for most of the outcomes was considered to be of high quality. For the outcomes of pain at 10 to 12 days, long-term effects, and serious complications, the evidence from the included studies was judged to be of moderate quality because of reporting bias, low sample size, and potential attrition bias.
Factors such as patients being recruited from different practice settings might have contributed to some heterogeneity among the trials. In addition, the duration of antibiotic therapy varied from seven to 14 days in the included trials. Most of the primary outcomes were measured within the first seven days of antibiotic therapy, but a longer duration of antibiotic therapy may have affected secondary outcomes.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
See theNNT.com's previous reviews of this topic:
Antibiotics for Acute Otitis Media, January 22, 2015
Antibiotics for Acute Otitis Media, August 22, 2010
Author
Maurice Selby, MD; Sigrid Wolfram, MDPublished/Updated
June 26, 2018References:
Erythromycin for Urgent Endoscopy After Upper Gastrointestinal Bleeding
Benefits in NNT
9
1 in 9 experienced decreased need for repeat endoscopy
4
1 in 4 experienced improved gastric mucosa visualization
Length of stay decreased by approximately 2 days
Harms in NNT
33
1 in 33 experienced increased gastrointestinal discomfort
View As:
Source
Rahman R, Nguyen DL, Sohail U, Almashhrawi AA, Ashraf I, Puli, SR, Bechtold ML. Pre-endoscopic erythromycin administration in upper gastrointestinal bleeding: an updated meta-analysis and systematic review. Annals of Gastroenterology. 2016 Jul-Sep; 29(3): 312-7.Study Population: 598 adults from 8 randomized controlled trials, primarily in ICU settings
Efficacy Endpoints
Need for 2nd endoscopy, hospital length of stay, units of packed red blood cells (PRBC) transfused, and visualization of gastric mucosaHarm Endpoints
Gastrointestinal discomfort, cardiac arrhythmiasNarrative
Upper gastrointestinal (GI) bleeding is a medical emergency that accounts for more than 500,000 hospital admissions each year and has a significant mortality of 5-10% with standard medical therapy.2 Urgent endoscopy with adequate visualization of the gastric mucosa is often essential for identifying the source of bleeding and treatment of these lesions. Traditionally, nasogastric lavage has been used to improve visualization, but this technique causes significant patient discomfort (pain and nausea) and risk (misplaced tube, risk of clot dislodgment). Erythromycin, a macrolide antibiotic, facilitates the motility of the gastric antrum and duodenum, thereby accelerating gastric emptying. For this reason, intravenous erythromycin has been proposed as a pre-endoscopy aid in visualization and treatment of upper GI bleeding.This meta-analysis includes eight trials (n = 598) involving adult patients with upper GI bleeding. All were randomized, controlled trials (RCTs) comparing erythromycin administration versus no erythromycin administration prior to endoscopy, but two of the studies were not blinded. All studies evaluated the primary endpoints of adequate visualization of the gastric mucosa and need for a second-look endoscopy.
Gastric visualization was assessed in each study as a dichotomous outcome and reported as adequate or inadequate visualization of gastric mucosa. Adequate gastric visualization was observed in 77% patients who received erythromycin before endoscopy and 51% who received no erythromycin or placebo (odds ratio [OR]: 4.14; 95% CI: 2.01-8.53). Erythromycin administration also resulted in a statistically significant decrease in the need for second-look endoscopy (OR 0.51; 95% CI: 0.34-0.77). Among secondary outcomes, there was a statistically significant decrease in hospital length of stay (5 trials, n = 375, mean difference [days]: MD -1.75; 95% CI: -2.43 to -1.06). No statistically significant difference was observed in duration of endoscopy (5 studies, n = 503) or need for emergent surgery (2 trials, n = 146). Administration of erythromycin was not associated with a statistically significant decrease in packed red blood cell (PRBC) transfusions (6 trials, n = 544, significant heterogeneity). However, a sensitivity analysis in which one study was removed showed significantly reduced heterogeneity and found that erythromycin administration decreased the number of blood transfusions compared to no erythromycin (mean difference [number of units]: -0.41; 95% CI: -0.82 to -0.01).
This meta-analysis did not explore harm outcomes, but several individual studies monitored for adverse effects. The medication was generally well tolerated, with only a few minor adverse reactions reported (GI discomfort).
Caveats
Two of the eight studies were not blinded, which introduced a risk of bias. However, a sensitivity analysis in which these two studies were removed the results remained similar in all measured outcomes. In addition, there was considerable variation of the erythromycin dose among the included trials, though studies have suggested an effect with as little as 70 mg of erythromycin,3 well below even the lowest doses used in these trials. As the authors point out, given the small number of patients in the included trials, mortality could not be fully assessed in this meta-analysis.Statistically significant heterogeneity was found in two outcomes (gastric visualization and procedural duration), but sensitivity analysis did not change the overall results. This heterogeneity may have been due to differences in study population, particularly in the percentage of the population with acute variceal bleeding. The authors suggest that erythromycin may be less effective in variceal bleeding, though this was not specifically addressed in the meta-analysis. Finally, three of the studies utilized nasogastric lavage, a significant potential confounder, before the administration of erythromycin. One prospective, randomized, multicenter trial4 compared intravenous erythromycin infusion without NG lavage, NG lavage without erythromycin, and the combination of both erythromycin and NG lavage. No statistically significant difference was seen among the three groups, suggesting that erythromycin and NG lavage may be similar in efficacy and that combining the two adds no significant benefit.
In light of these limitations, there appears to be a definite need for further research in this area. However, because of the reported benefits of improved visualization of gastric mucosa, decreased need for second-look endoscopy, and decreased hospital length of stay as well as low frequency of serious adverse effects, we recommend this intervention in adult patients with acute upper GI bleeding, particularly in those for whom nasogastric lavage prior to endoscopy is contraindicated or less desirable. We give it a rating of Green.
Author
Brian Resler, MD; Gary Green, MDSupervising Editor: Allan Wolfson, MD; Michael Ritchie, MD
Published/Updated
June 5, 2018References:
Combined Inhaled Short-Acting Beta2 Agonist and Anticholinergic Agents for Asthma
Benefits in NNT
16
1 in 16 avoided hospitalization
20
1 in 20 avoided relapse
Harms in NNT
36
1 in 36 had adverse events (tremors, agitation, or palpitations)
View As:
Source
Adams RJ, Fuhlbrigge A, Guilbert T, Lozano P, Martinez F. Inadequate use of asthma medication in the United States: results of the asthma in America national population survey. J Allergy Clin Immunol. 2002; 110(1):58–64.Study Population: Patients 16 years or older with asthma who presented to the emergency department (ED) with an asthma exacerbation
Efficacy Endpoints
Reduction in hospitalization, reduction in return visits to the EDHarm Endpoints
Increase in adverse events such as tremors, agitation, and palpitationsNarrative
In the United States, 9% of patients who present with an asthma exacerbation require hospitalization.1 The direct costs for asthma treatment in the United States, including prescriptions, hospitalizations, and clinic and ED visits, total approximately $5.1 billion annually.2 For years, the mainstay of treatment for acute asthma exacerbations has been short-acting beta2 agonists (SABAs), such as albuterol. Albuterol works primarily to promote bronchodilation. Additionally, short-acting anticholinergics, such as ipratropium (Atrovent), work to reduce bronchorrhea and have a milder impact on bronchodilation than SABAs. The combination of SABAs and short-acting anticholinergics has been studied and suggests a possible synergistic effect that lengthens symptom management and reduces the need for inpatient stays.This Cochrane review included 23 controlled or randomized trials with a total of 2,724 patients.3 Sixteen of these trials (2,120 participants) found that those receiving combined short-acting anticholinergic and SABA therapy had a lower rate of hospitalization than those receiving a SABA alone (relative risk [RR] = 0.72; 95% confidence interval [CI], 0.59 to 0.87). The estimated 65 fewer patients per 1,000 admitted to the hospital (166 vs. 231, respectively) translates to a number needed to treat (NNT) of 16. Five studies of 1,180 patients examined relapse, defined as a return to the ED with worsening symptoms within 24 hours. The results found that the combined therapy group was less likely to have a return visit (RR = 0.80; 95% CI, 0.66 to 0.98) with an estimated 50 fewer relapses per 1,000 patients (200 vs. 250, respectively) for an NNT of 20.3
In the 11 studies (n = 1,392 patients) that considered adverse events, patients who received combined therapy were more likely to experience adverse events than those in the group receiving SABA monotherapy (odds ratio = 2.03; 95% CI, 1.28 to 3.20).3 This translates to 28 more adverse events per 1,000 patients treated (131 vs. 103, respectively) for a number needed to harm of 36. These adverse events were found to be self-limiting and relatively minor, including (but not limited to) tremors, agitation, and palpitations.
Caveats
The overall quality of the studies included in these trials was considered to be low to moderate. Many sources of bias existed in these studies, including that 14 studies did not disclose their source of funding. Additionally, only one-half of the studies disclosed methods clearly enough to be considered a true randomized controlled trial. Many of the studies included also had small sample sizes.It is important to note that the benefit of a decrease in hospitalizations was found only in patients with severe asthma exacerbations, not those with mild to moderate exacerbations (test for difference between these subgroups found that P = .02). Regarding the decrease in relapse rates, there was heterogeneity in how this outcome was defined among the five studies analyzed (two studies defined relapse as return within 24 hours, one study as return within 48 hours, and two studies as return within two weeks). Furthermore, the authors were not able to examine potential differences in relapse rates among patients with mild, moderate, and severe exacerbations.
Fifteen studies compared hospitalization rates; of those, only five reported the criteria for admission. Interventions such as corticosteroid usage were clearly discussed in 13 studies; only in one-half did the study protocol dictate that corticosteroids be given to all participants, with the remainder of the trials leaving administration up to the physician. This may act as a confounder in these results.
Several studies attempted to evaluate improvement in forced expiratory volume in one second or percent increase above baseline, but the change in pulmonary function was not statistically significant between the two therapies and was deemed to be of low- or very-low-quality evidence in this review. However, 12 studies considered change in peak expiratory flow or percent improvement from baseline and found that patients given combined therapy did have an increase in both metrics compared with the SABA monotherapy group.
The description of adverse events across the studies was either too broad or not clearly defined, so it was not possible to extract specific adverse events for comparison.
Although the quality of evidence included in this review was low to moderate, the potential benefits of decreased hospitalization and relapse rates and the comparatively mild and limited adverse events suggest that patients presenting with acute asthma exacerbations are likely to benefit from combined short-acting anticholinergic and SABA therapy.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Dean A. Seehusen, MD, MPH; Daniel Runde, MDPublished/Updated
References:
Pregabalin (Lyrica) for Acute Fibromyalgia Pain
Benefits in NNT
11
1 in 11 experienced moderate to substantial symptom benefit after taking the medication
Harms in NNT
11
1 in 11 stopped taking the medication because of adverse effects
View As:
Source
Derry S, Cording M, Wiffen PJ, Law S, Phillips T, Moore RA. Pregabalin for pain in fibromyalgia in adults. Cochrane Database Syst Rev. 2016;(9):CD011790.Study Population: Adults with fibromyalgia
Efficacy Endpoints
At least a moderate to substantial symptom benefitHarm Endpoints
Withdrawal from trials because of adverse effectsNarrative
Fibromyalgia is a potentially debilitating disease characterized by widespread pain, fatigue, and sleep problems. In its most severe form, fibromyalgia can be incapacitating. High-quality studies analyzed in a Cochrane review consistently showed pregabalin (Lyrica) to have an effect on reducing pain in approximately 9% of patients with fibromyalgia, with a number needed to treat of 11.1 The studies showed a moderate to substantial symptom benefit in 331 (36%) of 932 patients receiving pregabalin compared with 251 (27%) of 937 patients receiving a placebo. Symptom improvement is an important endpoint that matters to patients and can have a positive effect on quality of life.1The studies also showed a number of adverse effects with pregabalin use, most commonly dizziness and somnolence. Adverse effects occurred in 17% of patients in the treatment group compared with 9% of patients in the placebo group. Patients had to discontinue taking the medication because of these adverse effects at about the same rate at which their symptoms improved, giving the medication a number needed to harm of 11. Given the benefit shown in multiple high-quality studies, we have labeled this treatment as green. However, patients should be made aware that they are equally as likely to experience intolerable adverse effects from the drug as they are to experience substantial benefit.
Caveats
The methodologic quality of the trials included in the Cochrane review was high. However, the studies did not assess the long-term tolerability of the medication. Most studies followed patients for 12 to 13 weeks (maximum of six months). This could be particularly important if the drug effectiveness decreases over time.One notable caveat is the potential for functional unblinding of patients allocated to the pregabalin arm in these trials. Although 9% of patients withdrew because of adverse effects, there may have been other patients who experienced common adverse effects not serious enough to cause termination of participation in the trials. For example, according to data from the U.S. Food and Drug Administration,2 somnolence occurred in 20% of patients treated with pregabalin, compared with 4% of participants receiving placebo. Similarly, 38% of patients allocated to receive pregabalin experienced dizziness compared with only 9% of patients receiving placebo. Patients experiencing adverse effects were more likely to have been in the treatment group, and unblinding may have been more likely in this group. There was no difference in the effectiveness of pregabalin with nighttime dosing or a divided twice-a-day regimen.
Pregabalin provides good pain relief to some patients with fibromyalgia but does not work for most persons. Physicians should consider switching patients to other medications after a trial of pregabalin, if the desired treatment effect has not been achieved.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Luke Donnelly, MD; Jaime Moran, MD; Mathew Foley, MDPublished/Updated
May 7, 2018References:
Qvar for Treatment of Chronic Asthma
Benefits in NNT
8
1 in 8 not receiving oral corticosteroids avoided an asthma exacerbation
2
1 in 2 receiving oral corticosteroids were able to discontinue oral corticosteroids
Harms in NNT
No harms were reported
View As:
Source
Adams NP, Bestall JB, Malouf R, Lasserson TJ, Jones PW. Inhaled beclomethasone versus placebo for chronic asthma. Cochrane Database Syst Rev. 2005;(1):CD002738.Study Population: Children and adults with a clinical diagnosis of chronic asthma
Efficacy Endpoints
Asthma exacerbation causing withdrawal from the trial; discontinuation of oral corticosteroidsHarm Endpoints
Oropharyngeal adverse effects such as dysphonia and oral candidiasisNarrative
During an asthma attack there is airway narrowing, wheezing, chest tightness, and coughing. Inhaled corticosteroids are used to improve pulmonary function and reduce the frequency of asthma attacks without exposing the patients to the adverse effects of systemic corticosteroids. Inhaled corticosteroids are the standard of care in preventing asthma attacks. Qvar is a new formulation of inhaled beclomethasone using a hydrofluoroalkane-134a propellant, in which the medicine is delivered in a solution. The smaller particle sizes in the hydrofluoroalkane-134a allows the medication to travel into the smaller parts of the lungs.The trials included in a Cochrane review studied the effectiveness of Qvar in two sets of patients with chronic asthma: those receiving oral corticosteroids and those not receiving them. These trials used different doses and delivery devices. In our review, we present only the combined data provided by the Cochrane review (any dose, any device).1
Other inhaled corticosteroids available are fluticasone (Flovent) and budesonide (Pulmicort). A different Cochrane review compared the effectiveness of three inhaled corticosteroids (fluticasone, beclomethasone, and budesonide) for treating patients with chronic asthma.2 Fluticasone at one half the daily dosage of beclomethasone or budesonide was at least as effective as the other two medications in improving airway opening. There were not enough data to compare the effectiveness of the three formulations in preventing acute asthma exacerbations. When given in the same dosage as the other two, fluticasone was associated with increased hoarseness, although it did not increase the risk of other adverse effects associated with corticosteroids such as oral thrush or sore throat.
In patients not receiving oral corticosteroids, the use of Qvar was associated with a lower risk of withdrawal from the trial because of asthma exacerbation (absolute risk reduction [ARR] = 12%; 95% confidence interval [CI], 9% to 16%; number needed to treat [NNT] = 8). Similarly, the use of Qvar allowed more patients to discontinue their oral corticosteroid (ARR = 46%; 95% CI, 35% to 56%; NNT = 2).
The risk of oral candidiasis and the overall oropharyngeal adverse effects were not statistically different between the groups in patients receiving oral corticosteroids.
Caveats
The source Cochrane review included two sets of populations among patients with chronic asthma, one group receiving oral corticosteroids and the other group not receiving them. The Cochrane review also measures the efficacy endpoints for various doses, delivery devices, and disease severities. For simplicity, we present only the data for any dose of Qvar irrespective of disease severity or the delivery device type.Most of the trials included in the review did not measure admission rates as an outcome. Most trials measured surrogate outcomes such as forced expiratory volume in one second, reduction in the frequency of use of rescue beta2 agonist, and the frequency of use of oral corticosteroids. These outcomes would only be meaningful if they could be translated into patient-centered outcomes such as reduction in emergency department visits or hospital admissions.
There was notable heterogeneity between the studies because of the different doses, disease severities, and delivery methods that were studied. Because of these differences, it is hard to combine the data for a singular conclusion.
It must be noted that the latest guidelines published by the National Heart, Lung, and Blood Institute also recommend Qvar for patients with chronic asthma.3
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Aditi Jayanth, MDPublished/Updated
April 19, 2018References:
Combination LABA Inhalers Compared with High-Dose Inhaled Steroids for Adults with Asthma
Benefits in NNT
73
1 in 73 avoided a mild asthma attack
Harms in NNT
1430
1 in 1,430 died because of an asthma attack
View As:
Source
Nelson HS, Weiss ST, Bleecker ER, Yancey SW, Dorinsky PM; SMART Study Group. The Salmeterol Multicenter Asthma Research Trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol [published correction appears in Chest. 2006;129(5):1393]. Chest. 2006;129(1):15–26.Study Population: Adult patients of any age and sex with known asthma of differing severities, already receiving an inhaled steroid
Efficacy Endpoints
Mild asthma exacerbation/attacks (defined as taking oral steroids), severe attacks (leading to hospitalization), life-threatening attacks (requiring mechanical ventilation), asthma-related deaths, and overall deathsHarm Endpoints
Death, asthma-related death, life-threatening asthma attackNarrative
Long-acting beta agonists (LABAs) relax smooth muscle in asthmatic lungs, potentially preventing attacks and improving symptoms. Although inhaled steroids are the accepted first-line controller, adding LABAs is common. LABAs increase severe attacks and asthma deaths when used alone.1 This summary examines the effectiveness and safety of LABAs when combined with steroids. There are many LABA and LABA/steroid comparisons. We focused on trials addressing a common and relevant clinical dilemma among patients with asthma: When taking an inhaled steroid and seeking better control, should the steroid dose be increased or should a LABA/steroid combination be used?The relevant Cochrane review examined 48 trials of more than 33,000 patients with asthma, assigning one group to a higher steroid dose and the other to a LABA/steroid. One in 73 patients in the LABA/steroid group avoided a mild attack (defined as requiring three to five days of oral steroids). However, adding a LABA did not reduce hospitalizations, deaths, or severe attacks. Moreover, for those with a 20% short-term risk of attack, one in 45 patients benefited; for those with a 1% risk of attack, one in 673 benefited. The more severe the asthma, the more likely the benefit. Adding a LABA also improved symptoms more than increasing the steroid dose, but at levels modest enough that asthma-related quality of life was unchanged.
Previous reviews have reached differing conclusions concerning safety. Two large reviews found that LABAs increase fatalities even when combined with steroids,2,3 suggesting approximately one in 1,400 patients will experience an asthma-related death. An updated 2014 review could neither confirm nor refute this danger, perhaps because of a lack of power. The authors' estimate for increased overall deaths was an odds ratio of 1.4 (0.6 to 3.4), suggesting the possibility of a 40% relative increase.4 An identical 40% increase was seen in the Salmeterol Multicenter Asthma Research Trial (SMART; odds ratio = 1.4, 0.7 to 2.9),1 although in both cases, the results were statistically insignificant.
The 2016 AUSTRI trial was one of five commissioned by the U.S. Food and Drug Administration to address the dangers of LABAs. AUSTRI compared LABA/steroid combinations with a steroid alone in nearly 12,000 adults with asthma. There were no asthma deaths in either group and just two life-threatening attacks, making it impossible to assess any differences in these outcomes. Based on similar hospitalization rates, the authors concluded that serious events were equal in both groups.5
Caveats
There are many reviews addressing LABA effectiveness and safety. For safety, we focus on a comparison we think reflects a common clinical choice, which is increasing the inhaled steroid vs. adding a LABA. However, other comparisons can be more relevant, for instance, when steroid doses are maximal or when other pharmacologic options with no safety problems (e.g., anticholinergics, leukotriene inhibitors) have been ineffective.6,7The length of trials complicates interpretation because they typically last just 12 or 26 weeks. The longer the use, the more patients may benefit. The Cochrane authors note that in the few longer-duration studies LABA benefits seemed to diminish or disappear after six months.
Symptom benefits with LABA/steroid therapy were small, typically 0.5 fewer pumps per day of a rescue inhaler and about 10% more symptom-free days, with no quality-of-life advantage. However, those with more symptoms may benefit more. They may also be at higher risk of a fatal attack, making LABAs tricky. The AUSTRI trial investigators' conclusion that LABA/steroid combinations are safe based on equivalent hospitalization rates is questionable. Hospitalizations are often unaffected by LABAs (see efficacy results above) and may not correlate with asthma deaths. Careful readers may also note that LABAs slightly reduced mild attacks; however, steroid doses were identical in the groups. Had doses been higher in the steroid group, the benefit for mild attacks might have disappeared. This is consistent with the finding that low-risk patients with asthma—the AUSTRI population—experience no meaningful benefit from LABAs. Future studies should focus on medium-risk and high-risk patients with asthma in whom fatal attacks are an uncommon but consistent threat, and for whom the potential benefit may be meaningful.
Finally, picking the most appropriate end point to review for numerical estimates is difficult, which is why we offer a range of values. All-cause mortality is a more patient-centered outcome than asthma-related deaths, but it is often not reported. We chose the Salpeter review as a primary source because it was the most transparent and conservative, and other reviews found the same or nearly identical point estimates.
We have chosen to designate this therapy red (no benefits) according to our rating system because of a small potential benefit of questionable clinical utility (avoiding a brief burst of oral steroids) and the possibility of a fatal harm. We considered black (harms greater than benefits); however, there is uncertainty about these fatal harms, and we hope physicians and patients can discuss these issues, share decisions, and come to their own conclusions.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Andrew Aherne, MD; John Kilpatrick, MDPublished/Updated
March 12, 2018References:
Anti-D (Rh factor) Administration During Late Pregnancy for the Prevention of Alloimmunization in At Risk (Rh Negative) Women
Benefits in NNT
No proven benefit
Harms in NNT
No serious harms
View As:
Efficacy Endpoints
Primary: Rhesus Rh D alloimmunization; Secondary: Kleihauer-Betke test, neonatal jaundiceHarm Endpoints
Anemia requiring transfusion, renal failure, disseminated intravascular coagulopathy (DIC), deathWho was studied: >4,500 Rh-negative un-sensitized women
Narrative
Alloimmunization occurs when a Rh negative (Rh-) mother develops anti-Rh antibodies after being exposed to Rh positive (Rh+) blood during pregnancy or in the immediate intra-partum period with a Rh+ fetus. Maternal Rh antibodies are capable of crossing the placenta, entering the fetal circulation of a subsequent pregnancy, and attacking fetal Rh+ red blood cells. This can lead to severe complications such as massive hemolysis, profound anemia, and high output fetal cardiac failure (hydrops fetalis).Anti-Rh immunoglobulin (aka Anti-Rh, Anti-Rh-D, Anti-D) quickly binds to fetal Rh+ red blood cells entering the maternal circulation and prevents them from sensitizing the mother’s immune system. Post-partum prophylaxis with Rh IgG has made the immune-mediated complications of Rhesus disease incredibly rare (Clarke and Hussey 1994; Crowther and Middleton 1997). Routine ante-partum administration of Rh immunoglobulin (Rhogam) to Rh- women, on the other hand, has the hypothesized role of preventing alloimmunization during pregnancy in the setting of occult fetal-maternal hemorrhage, thus preventing the aforementioned postpartum complications with Rh+ fetuses in future pregnancies.
This review included two relatively low quality trials involving over 4,500 Rh-negative unsensitized women. The studies examined the efficacy of IgG anti-D antibodies given at 28 and 34 weeks of gestation to prevent transplacental fetal blood exposure and maternal alloimmunization. Infusion of anti-D in Rh-negative pregnant women did not yield a significant change in alloimmunization rates during pregnancy, at delivery, or 12 months post-partum. Secondary outcomes showed a small but significant reduction in the chance of a positive Kleihauer-Betke test during pregnancy and at delivery. No differences were seen in neonatal jaundice. These findings were derived from low or very low quality data. The trials did not examine alloimmunization or other outcomes in future pregnancies.
Adverse effects of Rh immunoglobulin, including clinically-significant anemia, renal failure, pulmonary edema, DIC, and death, were not reported in these trials. Separate studies suggest adverse events to be exceedingly rare (MacKenzie et al 2004, e.g.).
Overall, this is a fairly well-performed systematic review and meta-analyses limited by mostly low quality evidence. Given the consistently demonstrated outcomes, potential patient-centered benefits, and lack of demonstrated or likely harm, we recommend consideration of the intervention (YELLOW), pending more definitive research.
Caveats
This latest Cochrane update by McBain et al from 2015 yielded one new trial in progress but no new published results to include since the prior article (Crowther and Middleton 2009). Therefore, data and conclusions remain unchanged. Methods were adjusted the prior publication to formally describe risk of bias using GRADE criteria.Maternal alloimmunization (the development of anti-Rh antibodies) has been widely accepted as a surrogate marker for potential fetal morbidity and mortality (Kim and Makar 2012). This is supported by population-based studies showing a marked decrease in rhesus disease since the routine administration of post-partum anti-D immunoglobulin (Clarke and Hussey 1994). The referenced Cochrane review did not show a significant difference in this outcome (low quality evidence).
The clinical accuracy and importance of the secondary outcome - a positive Kleihauer-Betke (measurement of fetal hemoglobin in maternal blood) - is much less certain (Duckett and Constantine 1997). The test is at best a weak proxy for potential alloimmunization in future pregnancies,and as such, is two steps removed from a patient-centered outcome.
The first trial included in the systematic review showed non-significant trends toward reduced alloimmunization using a 100 mcg dose. The second trial failed to show any benefit with a 50 mcg dose. This dose response mirrors what has been seen in postpartum administration. Reporting data for the higher-dose trials separately would yield a NNT for preventing alloimmunization of 213. This is roughly consistent with other non-randomized trials excluded by the Cochrane review but often cited as supporting data in obstetric guidelines. In general, the data for antepartum prophylaxis is less robust than that for postpartum women, with this systematic review showing only a trend toward benefit. Additionally, the included trials suffer from relatively poor quality, lacking placebo controls, absence of true blinding, and appropriate randomization.
Adverse events from Rho were not reported in the included trials of this review, but literature on the treatment of idiopathic thrombocytopenia (ITP) reports them to be exceedingly rare (Cooper 2009). It should be noted that the doses used in ITP (up to 200 mcg/kg) are much higher than those used for the prevention of rhesus disease, and many ITP regimens require multiple doses. It remains unclear how well estimates of harm equate in these two populations.
Given to Rh-negative women during the third trimester, anti-Rh IgG may have a role in reducing Rhesus disease in future pregnancies. Given the excellent safety profile of the intervention, the risk-to-benefit ratio suggests that the use of anti-Rh IgG in the third trimester may be a reasonable option in regions with sufficient resources and access to the drug. However, we believe that the lack of conclusive quantitative evidence has brought the cost effectiveness of unproven antepartum therapy into question, considering the cost of treatment may be on the order of $USD100-1,000. High-quality, randomized, placebo-controlled trials should be performed to assess whether either routine or event-based administration of anti-D immunoglobulin adds importantly to the routine use of the drug in the post-partum period.
Numbers:
Analysis I.2: Positive Kleihauer-Betke at birth of a Rh positive infant
Intervention 73/599 = 12.2%
Control 119/590 = 20.2%
RRR = 39.6%
ARR = 7.98%
NNT = 12.5
X100/60 (for all pregnancies not just Rh+) = 20.8
Analysis I.4: Immunization in pregnancy *NON SIGNIFICANT
Intervention 5/1879 = 0.3%
Control 13/2023 = 0.6%
RRR = 58.6%
ARR = 0.4%
NNT = 265.6
Analysis I.5: Immunization after birth of a Rh positive infant *NON SIGNIFICANT
Intervention 5/1112 = 0.4%
Control 13/1185 = 1.1%
RRR = 59.0%
ARR = 0.6%
NNT = 154.5
X100/60 = 257.4
Analysis I.6: Immunization at 2-12 months *NON SIGNIFICANT
Intervention 6/985 = 0.6%
Control 16/1063 = 1.5%
RRR = 59.5%
ARR = 0.9%
NNT = 111.6
X100/60 = 186.0
“This result became statistically significant when calculated as a risk difference (RD) across the two randomised controlled trials (RD -0.01, 95% CI -0.01 to 0.00).”
Analysis I.9: Neonatal Jaundice *NON SIGNIFICANT
Intervention 1/927 = 0.1%
Control 4/955 = 0.4%
RRR = 74.2%
ARR = 0.3%
NNT = 321.6
Author
Gary Green, MDPublished/Updated
February 8, 2018References:
Topical Capsaicin for Treatment of Chronic Neuropathic Pain in Adults
Benefits in NNT
12
1 in 12 patients reported postherpetic pain reduction of at least 50% more than the control group by follow-up within eight weeks
10
1 in 10 patients reported postherpetic pain reduction of at least 30% more than the control group by follow-up within eight and 12 weeks
11
1 in 11 patients reported human immunodeficiency virus neuropathy pain reduction of at least 30% more than the control group at follow-up within 12 weeks
Harms in NNT
3
None of the participants withdrew from care because of adverse reactions
1 in 3 patients in the active treatment group experienced transitory pain at the application site
View As:
Source
Derry S, Rice AS, Cole P, Tan T, Moore RA. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database Syst Rev. 2017;(1):CD007393.Study Population: Patients 16 years or older with neuropathic pain of any origin for more than 12 weeks
Efficacy Endpoints
Patients reported postherpetic pain reduction of at least 50% more than the control group by follow-up within eight weeks; patients reported postherpetic pain reduction of at least 30% more than the control group by follow-up within eight and 12 weeks; and patients reported human immunodeficiency virus neuropathy pain reduction of at least 30% more than the control group at follow-up within 12 weeks.Harm Endpoints
No additional patients withdrew from care because of adverse reactions. One in three patients in the active treatment group experienced transitory pain at the application site.Narrative
Chronic neuropathic pain is a common, sometimes debilitating, condition that can result from a multitude of origins. Currently, pregabalin (Lyrica) is the only approved medication option. Physicians commonly prescribe off-label use of selective serotonin reuptake inhibitors, gabapentin (Neurontin), tricyclic antidepressants, and antiepileptics. This review assesses the effectiveness and duration of a single application of high-concentration (8%) topical capsaicin for pain relief in patients with neuropathic pain lasting more than eight weeks.A 2017 Cochrane review of eight double-blind, randomized controlled studies included 2,488 patients, and compared the effect of a single topical application of high-concentration (8%) capsaicin for long-term neuropathic pain relief compared with a control group.1 A 0.04% concentration capsaicin control was used in all studies to blind participants to the burning and erythema experienced after application. Pain was not assessed in the first posttreatment week because capsaicin preparations often cause localized pain at the application site for several days.
For patients with postherpetic neuropathic pain, topical capsaicin provided moderate pain relief (30% to 50% reduction from baseline) with a number needed to treat (NNT) of 12 (29% vs. 20% in high-concentration application and control groups, respectively) to reduce pain by 50% at follow-up within eight weeks, and an NNT of 10 (43% vs. 34% in high-concentration application and control groups, respectively) to reduce pain by at least 30% within eight weeks. Three studies also looked at 30% pain reduction within 12 weeks and found a similar benefit, with an NNT of 10 (46% vs 37% in high-concentration application and control groups, respectively).
This review also included two studies that found at least a 30% reduction in pain in patients with human immunodeficiency virus neuropathy pain treated with a single application of high-concentration capsaicin with an NNT of 11 at follow-up within 12 weeks (39% vs. 30% in high-concentration application and control groups, respectively). These studies further differentiated effects on pain with a 30-minute total application time vs. 60-minute total application time, which did not show a meaningful difference in outcomes. The 30-minute application of high-concentration capsaicin had a relative risk (RR) of 2.95 (95% confidence interval [CI], 0.73 to 11.88) compared with the 60-minute application of high-concentration capsaicin, which had an RR of 1.34 (95% CI, 1.03 to 1.75).
Effects on diabetic peripheral neuropathy and neuropathic pain following inguinal herniorrhaphy were also reviewed; however, data in these cohorts were sparse. High-concentration topical capsaicin did not improve these conditions.
The most common adverse events were erythema and pain over the application site compared with the control group; however, these effects were transitory and minimal for almost all participants. There were 12 withdrawals because of adverse reactions in 1,507 participants treated with high-concentration capsaicin compared with nine withdrawals in 980 participants in the control group (number needed to harm was not calculated).
Caveats
Although the studies included 2,488 participants, the quality of evidence for each trial was very low to moderate given wide CIs, sparse data, and susceptibility to publication bias. About 50% of patients (n = 1,272) in this meta-analysis had postherpetic neuropathy. Improved heterogeneity in study populations to include more participants with other etiologies of neuropathic pain would allow for data to be generalized over a greater population. Additionally, better quality studies are needed to demonstrate definitive improvement.Based on low-quality evidence, high-concentration capsaicin patches reduce neuropathic pain from herpes and human immunodeficiency virus infection by 30% to 50% compared with the low-concentration capsaicin control group.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Paul Crawford, MD and You Xu, MDPublished/Updated
January 11, 2018
Stents for Stable Coronary Artery Disease
Benefits in NNT
No benefit in patients with stable nonacute coronary artery disease in the five-year follow-up period
Harms in NNT
50
1 in 50 experienced a serious complication such as death, stroke, myocardial infarction, arrhythmia, hemorrhage
View As:
Source
Stergiopoulos K, Boden WE, Hartigan P, et al. Percutaneous coronary intervention outcomes in patients with stable obstructive coronary artery disease and myocardial ischemia: a collaborative meta-analysis of contemporary randomized clinical trials. JAMA Intern Med. 2014;174(2):232–240.Study Population: Adults, typically 55 to 65 years of age, mostly men, with stable nonacute coronary artery disease (not actively having ischemia or a myocardial infarction) diagnosed by abnormal exercise stress testing, nuclear or echocardiographic stress imaging, or fractional flow reserve (FFR)
Efficacy Endpoints
Death, nonfatal myocardial infarction, angina symptomsHarm Endpoints
Death, stroke, myocardial infarction, arrhythmia, hemorrhage, contrast media reactionNarrative
Percutaneous coronary intervention (PCI), generally with stenting, is commonly used to open occluded coronary arteries. This review summarizes the evidence on the benefits of this procedure in patients diagnosed with stable nonacute coronary artery disease (without acute ischemia or myocardial infarction). This diagnosis is usually made after an abnormal exercise stress test, or nuclear or echocardiographic stress imaging.The benefits of PCI in patients with acute ischemia (myocardial infarction) will be discussed in a future review.
The meta-analysis discussed here analyzed data on 5,286 patients from five trials. Patients with stable obstructive coronary artery disease were randomized to receive PCI and medical treatment or medical treatment alone. All patients had a previous positive stress test or abnormal FFR, which is a test commonly used in the cardiac catheterization laboratory to assess the significance of a coronary stenosis by passing a thin wire through the occlusion and measuring any drop in pressure. FFR allows for making treatment decisions based on impairment of blood flow, not just visualizing the stenosis. Not all patients included in the meta-analysis had FFR measured.
After an average of five years of follow-up, coronary stenting for stable nonacute coronary artery disease did not lead to statistically significant changes in the risk of death (7.3% in medical treatment group vs. 6.5% in PCI group; odds ratio [OR] = 0.90; 95% confidence interval [CI], 0.71 to 1.16; P = .42); nonfatal myocardial infarction (7.6% in medical treatment group vs. 9.3% in PCI group; OR = 1.24; 95% CI, 0.99 to 1.56; P = .06); unplanned revascularization (28.5% in medical treatment group vs. 18.3% in PCI group; OR = 0.64; 95% CI, 0.35 to 1.17; P = .14); or angina (23.3% in medical treatment group vs. 20.3% in PCI group; OR = 0.91; 95% CI, 0.57 to 1.44; P = .67).1
Cardiac angiography is an invasive procedure with potentially serious complications including death (0.11%), myocardial infarction (0.05%), stroke (0.2%), arrhythmia (0.4%), vascular complications (0.4%), contrast media reaction (0.4%), hemodynamic complications (0.3%), and other (0.3%).2, 3, 4 Subjecting patients to a 2% risk of a major complication may not be justified in the absence of clear benefits. None of the trials included in the meta-analysis reported the complications associated with PCI. Therefore, we calculated the number needed to harm based on the data reported in the American Heart Association/American College of Cardiology (AHA/ACC) guidelines, which used the Society for Cardiac Angiography and Interventions registry database as well as the ACC's National Cardiovascular Data Registry (CathPCI Registry) as their sources.2,3 Future randomized controlled trials reporting complications of PCI procedures may provide a more accurate estimate of risk.
Caveats
Most published trials fail to show a benefit from coronary stenting in patients with stable obstructive coronary artery disease. However, stent technology has evolved significantly in recent years, and it is possible that newer-generation drug-eluting stents may offer different results.It has been suggested that the following patient subgroups might derive net benefit from PCI:
Higher-risk patients with moderate to severe objective myocardial ischemia (without myocardial infarction). A more recent meta-analysis5 reported fewer cardiac deaths with PCI compared with medical treatment (hazard ratio = 0.52; 95% CI, 0.30 to 0.92; P = .02) in the context of objective ischemia, contradicting the previous data.1 Of the three trials included, the bulk of patients (888 out of 1,557) came from the FAME 2 trial (Fractional Flow Reserve versus Angiography for Multivessel Evaluation 2), which did not demonstrate a reduction in mortality or myocardial infarction with PCI. The mortality benefit reported was likely driven by the SWISSI 2 trial (Swiss Interventional Study on Silent Ischemia Type II), which reviewed treatment of silent ischemia after ST segment elevation myocardial infarction. Patients included in SWISSI 2 were different from the patients under consideration here, who were higher risk (not stable) having demonstrated a tendency for plaque rupture and myocardial infarction.
Patients with angina symptoms despite optimal medical therapy. In patients on maximal antianginal drug therapy with continuing life-limiting angina, PCI of the stenosis might be beneficial for alleviating these symptoms as a last resort. However, unlike the objective end points discussed above, anginal severity is subjective and, as such, any improvement might be because of the placebo effect from the procedure itself. The recently published ORBITA study (Optimal Randomized Blinded Investigation with Optimal Medical Therapy of Angioplasty in Stable Angina), although small, reported that PCI did not significantly change anginal severity or frequency in medically optimized patients when compared with a sham procedure.6 The importance of an optimal drug regimen cannot be overstated. Benefits of PCI appear to be minimal in patients who are receiving optimal current medical management.7
The ISCHEMIA trial (International Study of Comparative Health Effectiveness with Medical and Invasive Approaches),8 which is currently recruiting, will further inform the debate. This trial compares an initial strategy of medical treatment, with catheterization reserved for patients whose symptoms do not improve with this intervention, to an initial invasive strategy. The results of this open-label trial are expected in December 2018.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Andrew Apps, BMBCh, MRCP and Aseem Malhotra, MBChB, MRCPPublished/Updated
January 8, 2018References:
Acetazolamide for Prevention of Acute Mountain Sickness (AMS)
Benefits in NNT
8
1 in 8 patients had prevention of AMS at high altitude 24 hours after arrival
Harms in NNT
There was no evidence of life threatening side effects.
2.4
NNH 2.4 for paresthesias in those prescribed acetazolamide
View As:
Source
Nieto Estrada VH, Molano Franco D, Medina RD, Gonzalez Garay AG, Martí-Carvajal AJ, Arevalo-Rodriguez I. Interventions for preventing high altitude illness: Part 1. Commonly-used classes of drugs. Cochrane Database of Systematic Reviews 2017, Issue 6. Art. No.: CD009761. DOI: 10.1002/14651858.CD009761.pub2.Study Population: Approximately 2,300 patients across 16 studies with recent ascent to altitude at 4,001-5,000 meters
Efficacy Endpoints
Development of AMS, defined by Lake Louise score or other study-specific criteria from arrival to 24 hours laterHarm Endpoints
Report of paresthesias from arrival to 24 hours laterNarrative
Above 2,500 meters, the partial pressure of oxygen in the atmosphere drops, requiring the human body to adapt. In the acute stage, these adaptations include hyperventilation, tachycardia and an increase in blood pressure. If ascent occurs rapidly, and physiologic adaptation is insufficient, Acute Mountain Sickness (AMS) can result. The symptoms AMS include headache, nausea, anorexia, vomiting, lightheadedness, insomnia and fatigue. If the condition persists, in the right individual and the right circumstances, these symptoms can progress to potentially fatal High Altitude Cerebral Edema (HACE) and/or High Altitude Pulmonary Edema (HAPE).Prophylaxis is often attempted using the carbonic anhydrase inhibitor acetazolamide. Although the exact mechanism of action is unclear, one theory is that acetazolamide inhibits carbonic anhydrase in the kidneys, increasing bicarbonate excretion, resulting in a metabolic acidosis that offsets the hyperventilation-induced alkalosis experienced at altitude. However, acetazolamide likely has multiple effects throughout the body that help to prevent AMS. Both patients and providers should be aware that relief of AMS can come at the expense of side effects, most commonly paresthesias, but also polyuria, rash, and dysgeusia.
It should also be noted that the doses of acetazolamide used in these studies are higher than doses that have been shown to be as effective at preventing AMS. The incidence of adverse effects also increases as the dose of acetazolamide increases. This review may therefore overstate the risk of paresthesias. Although the frequency of paresthesias when taking this medication is high, this side effect is not life threatening and there is clear benefit in the reduction of debilitating AMS that may progress to life-threatening HACE if untreated. For this reason, we have given this treatment a rating of green.
Caveats
The trials included in this systematic review were heterogeneous in regard to number of days before starting the ascent that the intervention was started, method of ascent (trekking, gondola, etc.), altitude ascended to, dose of acetazolamide provided, and predisposition of patients to AMS as per prior history and comorbidities (asthma, chronic obstructive pulmonary disease, diabetes, etc.). Nonetheless, they were deemed homogeneous enough for comparison after a statistical analysis of heterogeneity showed differences between studies to be non-significant. Of note, the authors of the review point out that 70% of studies do not clearly describe the method of randomization. Furthermore, this review included studies in which patients were evaluated for symptoms up to 24 hours after ascent. Onset of symptoms (and side effects of medication) could first be noted beyond 24 hours, usually within the first few days. Treatment with acetazolamide would likely extend past 24 hours, further complicating the picture and possibly the likelihood of side effects.Studies that evaluated paresthesias were frequently unclear in specifying if symptoms were elicited or spontaneously reported. Drop-out rates for acetazolamide arms specifically secondary to paresthesias are difficult to assess from the data gathered. It seems that most of the included studies did not report on how many people dropped out secondary to paresthesias alone. Authors of this review attempted to use an intention-to-treat analysis in order to address any break from experimental protocol secondary to any side effects of medication.
The definition of AMS was also variable. Authors note that 35% of trials use the Lake Louise score, which is specifically designed to evaluate AMS. In 30% of the studies the categorization of AMS was “unclear,” and in the remainder the diagnostic criteria were not commented on. The variability of the definition of AMS across studies could significantly affect the interpretation of data.
Numbers:
Benefit from arrival to 24 hours later (prevention of AMS):
2301 participants over 16 studies
Placebo group: 252/1046 (24.1%)
Acetazolamide group: 139/1255 (11.1%)
RR for acetazolamide group: 0.47 (95% CI, 0.39 to 0.56)
Absolute risk reduction: 13%
NNT: 7.7
Harm Outcome (paresthesias experienced from arrival to 24 hours later):
789 participants over 5 studies
Placebo group risk: 32/351 (9.1%)
Acetazolamide group: 222/438 (50.6%)
RR for acetazolamide group: 5.53 (95% CI, 2.81 to 10.88)
Absolute risk increase: 41%
NNH: 2.4
Author
Kyle Kelson, MD and Carlo Canepa, MDPublished/Updated
November 16, 2017References:
Statins in Persons at Low Risk of Cardiovascular Disease
Benefits in NNT
No statistically significant mortality benefit
217
1 in 217 avoided a nonfatal heart attack (myocardial infarction)
313
1 in 313 avoided a nonfatal stroke
Harms in NNT
21
1 in 21 experienced pain from muscle damage
204
1 in 204 developed diabetes mellitus
View As:
Efficacy Endpoints
Death, heart attack (myocardial infarction), strokeHarm Endpoints
New-onset diabetes mellitus, muscle symptomsNarrative
Statins may prevent clotting events by reducing cholesterol and arterial plaque in blood vessels. Vascular diseases are important causes of death and disability, and there is evidence that statins reduce mortality, heart attacks, and strokes in those at high risk (20% or higher 10-year risk of cardiovascular disease).1 Whether the drugs should be used for persons at lower risk, however, is controversial.2The summary of the 2012 Cholesterol Treatment Trialists (CTT) meta-analysis is unique because it shows trial results according to baseline risk.3 For nonfatal stroke and nonfatal heart attack, the CTT meta-analysis does not show outcomes based on risk and does not report them separately from fatal events. Therefore, the U.S. Preventive Services Task Force (USPSTF) review of statins for primary prevention is used for these end points.4 The occurrence of adverse effects associated with statins is summarized from three sources: the USPSTF summary of serious adverse events (generally, all illnesses and those requiring hospitalization),4 one randomized trial of statin-induced muscle damage,5 and the largest meta-analysis of statin-induced diabetes.6
The CTT meta-analysis, which included 22 trials with more than 130,000 patients, showed no statistically significant mortality benefit from statins in the two low-risk groups (lower than 10% and lower than 20% 10-year risk), separately and combined.3,7 Conversely, the USPSTF, pooling data from 15 trials with more than 70,000 patients, found that 0.4% fewer patients taking a statin died than patients taking placebo (number needed to treat [NNT] = 250).4 Importantly, some of the trials in the USPSTF analysis included high-risk patients or patients with cardiovascular disease.
The USPSTF found that statins have a 0.46% absolute benefit (NNT = 217) for nonfatal heart attacks.4 This NNT is probably artificially improved (lowered) by the predominance of events that occurred in high-risk individuals. In trials, however, 30% to 40% fewer heart attacks occurred in statin groups compared with placebo groups, suggesting that the benefit is reliable and can therefore be accepted as the best available estimate. Nonfatal strokes occurred in 0.32% fewer patients taking statins (NNT = 313), with the same caveats.4
Importantly, despite the small reductions in nonfatal heart attacks and strokes, statins were not associated with a reduction in serious illness overall (relative risk = 0.99; 95% confidence interval, 0.94 to 1.04).4 Adverse events from statin use include myalgia and new-onset diabetes at a rate of 4.8% (number needed to harm [NNH] = 21) and 0.49% over five years (NNH = 204), respectively.5,6 Other adverse events, although often reported, have not been well studied.
Caveats
There is controversy about whether statins reduce all-cause mortality in low-risk persons—analyses finding a mortality benefit universally included some high-risk patients. Although statins provide a significant reduction in mortality in high-risk groups, this benefit has not been shown in lower-risk groups. This could be because of underpowered trials (i.e., insufficient numbers of low-risk patients included). If so, any mortality benefit would be small and would result in a very large NNT.The importance of the adverse effects of statins, although historically minimized, is becoming increasingly apparent.8 The Effect of Statins on Skeletal Muscle Function and Performance (STOMP) trial found that statin-induced myalgias are relatively common at 4.8%.5 The trial also found that patients taking statins commonly have increased creatine kinase enzyme levels, indicating some degree of muscle damage.The 4.8% incidence of statin-induced myalgias is almost certainly an underestimation because the STOMP trial enrolled patients for six months, which is one-tenth of the median duration of trials testing benefits. Because it is unclear if the incidence of statin-related muscle symptoms increases with time, the results cannot be extrapolated forward. Clinicians and patients should be aware that the true incidence of statin-related muscle symptoms is likely higher than reported in the STOMP trial. It should also be noted that only one of the 42 trials included in a meta-analysis addressing statin-induced muscle problems prospectively inquired about muscle symptoms.9
The USPSTF review found no increase in the risk of new-onset diabetes associated with statin therapy for primary prevention.4 However, the best predictor of developing statin-induced diabetes is diabetes risk, not cardiovascular risk.10 Therefore, analyzing exclusively primary prevention data may have underpowered the analysis. A 2010 meta-analysis of 13 trials with more than 90,000 patients, which is broadly cited as the most complete assessment of statin-induced diabetes, shows a 9% relative increase (not far from the USPSTF’s result of 5%) in diabetes risk.6 The absolute increase was 0.098% per year of statin exposure.6 This becomes 0.49% at five years, although the number will vary with diabetes risk, just as statin benefits vary with cardiovascular risk.
In summary, studies have found no significant overall mortality benefit with statin therapy in low-risk patients, as well as no reduction in the risk of serious illness overall and very small benefits for nonfatal heart attack and stroke. Statins also appear to cause diabetes. Although this is uncommon, diabetes may occur more often than the prevention of a heart attack or stroke in patients taking statins. It appears that the existing evidence is in disagreement that statins should be used for patients with a 10-year cardiovascular risk below 20%.4,11,12 With no mortality benefit, no reduction in serious illness, an approximately 1% chance of avoiding a nonfatal heart attack or stroke, a similar or greater chance of developing diabetes, and a one in 21 chance of muscle damage, it seems wiser to focus on lifestyle changes (such as adopting a Mediterranean diet, exercising, and not smoking) instead of cholesterol drugs in low-risk patients. These individuals should be informed of the known risks and benefits of statins, and the decision to start statin therapy should be shared by the patient and physician, rather than imposed by guidelines.
We have categorized statins for low-risk patients as red, or not recommended, based on certain value judgments. Statin studies, mostly industry sponsored, used methods such as run-out phases, and the raw trial data continue to be withheld by manufacturers despite many requests by independent groups.13 Thus, it is reasonable to assume that the reported benefits represent a best-case, whereas harms are most likely underestimated. In addition, although statin-induced muscle symptoms are at least five times more likely than any benefit, this is typically reversible. The decision not to categorize statins for low-risk patients as black, or harms greater than benefits, is based on value judgments about this compared with cardiovascular events. This decision becomes trickier when considering the additional burden of statin-induced diabetes. One large, high-quality trial did not find an increase in diabetes risk.14 However, originally unpublished results from the Stroke Prevention by Aggressive Reduction in Cholesterol Levels trial failed to disclose that the NNH for new-onset diabetes was just 38 in patients treated with atorvastatin (Lipitor), 80 mg, compared with placebo.15
Finally, revascularization was not used as an end point because therapeutic decisions are nonstandardized, patients in statin trials are functionally unblinded by lowered low-density lipoprotein cholesterol levels, and the procedure does not save lives or prevent heart attacks in stable patients.16
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
See theNNT.com's previous reviews of this topic:
Statin Drugs Given for 5 Years for Heart Disease Prevention (Without Known Heart Disease), January 10, 2015
Statin Drugs Given for 5 Years for Heart Disease Prevention (Without Known Heart Disease), November 2, 2013
Author
John Abramson, MD, MScPublished/Updated
November 8, 2017References:
Varenicline for Smoking Cessation
Benefits in NNT
11
1 in 11 stopped smoking
Harms in NNT
5
1 in 5 had nausea
15
1 in 15 had abnormal dreams
23
1 in 23 had insomnia
50
1 in 50 had headache
143
1 in 143 had serious adverse events (infections, cancers, injuries)
View As:
Source
Cahill K, Lindson-Hawley N, Thomas KH, Fanshawe TR, Lancaster T. Nicotine receptor partial agonists for smoking cessation. Cochrane Database Syst Rev. 2016;(5):CD006103.Study Population: Adult smokers willing to attempt quitting smoking
Efficacy Endpoints
Smoking cessation at least six months from the start of interventionHarm Endpoints
Medication side effects or adverse eventsNarrative
Worldwide, smoking remains the main preventable cause of morbidity and premature death. Dependence on nicotine reflects the effects of the drug on neuronal receptors in the brain, which stimulate the release of dopamine. Varenicline (Chantix) is a medication that aids in smoking cessation by acting as a nicotine receptor partial agonist, and is one of seven first-line medications recommended to increase long-term smoking abstinence.1 The authors of a systematic Cochrane review performed a meta-analysis examining 39 randomized trials (N = 11,801) that tested varenicline in a variety of populations and settings, and against various comparators.2 Trials included in the review variably enrolled adult smokers with the goal of cessation for at least six months.Varenicline adminstered at the standard dosage (1 mg twice daily) more than doubled the chances of quitting compared with placebo, with a pooled risk ratio [RR] of 2.24 (95% confidence interval [CI], 2.06 to 2.43; 27 trials, N = 12,625; high-quality evidence). Varenicline at a reduced dosage (0.5 mg twice daily) remained effective with an RR of 2.08 (95% CI, 1.56 to 2.78; four trials, N = 1,266), a cessation rate that is similar to varenicline vs. bupropion (Zyban), with an RR of 1.39 (95% CI, 1.25 to 1.54; five trials, N = 5,877; high-quality evidence), and nicotine replacement therapy, with an RR of 1.25 (95% CI, 1.14 to 1.37; eight trials, N = 6,624; moderate-quality evidence). Based on this evidence and using advanced statistical modeling, the authors calculated the number needed to treat (NNT) with varenicline as 11 (95% CI, 9 to 13). Using the compiled data and absolute risk reduction without advanced statistics gives an NNT for varenicline vs. placebo, bupropion, and nicotine replacement therapy of 6, 15, and 20, respectively.
The predominant adverse event was mild to moderate nausea subsiding over time, with rates between 6% and 51%. Rates of serious adverse events were noted to be about 25% higher in those receiving varenicline compared with the control groups, with an RR of 1.25 (95% CI, 1.04 to 1.49; 29 studies, N = 15,370; I2 = 0%), and included comorbidities such as infections, cancers, and injuries, most of which were considered to be unrelated to the treatment. The number needed to harm (NNH) for one serious adverse event as calculated by the authors using advanced statistical modeling is 143 (95% CI, 74 to 556). Without advanced statistical methods and using the compiled data resulted in an NNH of 165.
Caveats
In 2009, the U.S. Food and Drug Administration (FDA) added a black-box warning to varenicline because of post-marketing safety reports of an association between varenicline and psychiatric adverse events including depressed mood, agitation, and suicidal behavior/ideation.3 The FDA required Pfizer, the manufacturer of Chantix, to conduct a clinical trial to evaluate the risk of neuropsychiatric adverse events, and found the risk to be lower than previously suspected. As a result, the FDA removed the black-box warning for serious mental health side effects from the Chantix drug label on December 16, 2016.In 2011, the FDA advised that varenicline might slightly increase cardiovascular events in persons with cardiovascular disease.3 Results are pending from a large trial that should give more definitive data on this issue. Should data become available suggesting a significant increase in major adverse cardiac events, it would necessitate revisiting this topic and potentially changing the recommendation. A 2015 update from the FDA added a warning that varenicline may change the way people react to alcohol (e.g., possible increased drunkenness, unusual behavior, memory lapse), as well as rare accounts of seizures with treatment. These warnings were added after a review of the FDA Adverse Event Reporting System and a case series submitted by Pfizer. The actual NNT and NNH vary depending on how the calculation is performed. However, calculations without advanced statistical modeling are close to those reported by the authors, and show varenicline to be beneficial.
The serious adverse events data do not differentiate between events caused by treatment vs. comorbidities. Also, losses to follow-up were higher in the control groups (mean of 28.4%) than in the treatment groups (mean of 23.8%) of most of the studies (25 of 29), which may underestimate serious adverse event rates in the control groups. Thus, serious adverse event rates and associated NNH attributable to varenicline are likely overestimated.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Paul Crawford, MD, PhD and Daniel Cieslak, MDPublished/Updated
September 1, 2017References:
Antipsychotics for Fibromyalgia in Adults
Benefits in NNT
8
1 in 8 had at least a 30% reduction in pain
4
1 in 4 had a reduction in sleep problems
6
1 in 6 had a reduction in depressed mood
5
1 in 5 had a clinically significant improvement in quality of life
Harms in NNT
12
1 in 12 gained 11 lb (5 kg) or more
View As:
Source
Walitt B, Klose P, Üçeyler N, Phillips T, Häuser W. Antipsychotics for fibromyalgia in adults. Cochrane Database Syst Rev. 2016;(6):CD011804Study Population: Adults with fibromyalgia
Efficacy Endpoints
At least 30% reduction in pain; reduction in depressed mood; reduction in sleep problems; clinically significant improvement in quality of lifeHarm Endpoints
Any adverse event including weight gain and participants dropping out because of an adverse effectNarrative
Fibromyalgia is characterized by chronic, widespread musculoskeletal pain, often associated with fatigue, memory problems, and sleep disturbances.1 However, ongoing research suggests it is a disorder of pain regulation or central sensitization.2 Up to 70% of patients with fibromyalgia also meet criteria for depression or anxiety, so medications for these conditions are often used to treat fibromyalgia.This Cochrane review evaluated the evidence available for the use of antipsychotics, specifically quetiapine (Seroquel), to treat fibromyalgia.3 A total of four studies with 296 participants were reviewed. Three studies compared the use of quetiapine at bedtime with placebo (n = 206), and the fourth study (n = 90) compared quetiapine with amitriptyline. For the primary measured outcome of at least 50% reduction in pain, quetiapine was not statistically more effective than the control. For the secondary outcome, reduction of pain by at least 30%, two studies (n = 155) found a statistically significant benefit. Twenty of 82 participants (24.4%) receiving quetiapine and eight of 73 participants (11.0%) receiving a placebo reported pain relief of at least 30% (risk difference [RD] = 0.12%; 95% confidence interval [CI], 0.00 to 0.23; P = .04). The number needed to treat (NNT) for an additional person to benefit was 8 (95% CI, 5 to 100).
In two studies (n = 87), there was evidence that quetiapine was superior to placebo in reducing sleep problems, another secondary end point (standardized mean difference [SMD] = −0.67; 95% CI, −1.10 to −0.23; P = .003; NNT = 4).
Three studies (n = 207) were evaluated in an analysis of depression, a secondary end point. Quetiapine was superior to placebo in reducing depressed mood (SMD = −0.39; 95% CI, −0.74 to −0.04; P = .03; NNT = 6).
The final secondary end point was participant-reported improvement of health-related quality of life in the Fibromyalgia Impact Questionnaire (FIQ). Three studies (n = 206) were analyzed. The available evidence suggested that quetiapine was superior to placebo. Fifty-three of 107 participants (49.5%) in the quetiapine group and 32 of 99 participants (32.3%) in the placebo group reported a reduction of 14% or more of the FIQ total score (RD = 0.18; 95% CI, 0.05 to 0.31; P = .008; NNT = 5). A change in FIQ score of 14% has previously been defined as the minimal difference that is clinically relevant.4
Three studies with 206 participants were analyzed for safety and tolerability. The analysis showed very-low-quality evidence for no statistically significant difference between quetiapine and placebo in regard to withdrawal because of adverse events (RD = 0.10; 95% CI, −0.06 to 0.27; P = .21) and serious adverse events (RD = 0.00; 95% CI, −0.03 to 0.03; P = .96).
Two studies (n = 155) showed a statistically significant difference between quetiapine and placebo in weight gain. Seven of the 82 participants (8.5%) receiving quetiapine and none of the 73 participants receiving placebo reported a substantial weight gain (more than 11 lb [5 kg]; RD = 0.08; 95% CI, 0.02 to 0.15; P = .01.) The number needed to treat to harm was 12.
Caveats
The evidence reviewed was considered to be of generally low quality because of small sample sizes, imprecise results, and bias risk from limited study designs. Intended subgroup analyses, such as those with and without depression, could not be completed because of an insufficient number of studies. Quality of evidence was also downgraded because of indirectness (exclusions of patients with disease) and imprecision (fewer than 400 patients analyzed). Larger and more methodologically sound studies will be needed to verify the conclusions.Quetiapine was the only antipsychotic the authors found that was studied in adults with fibromyalgia. Doses of quetiapine ranged from 50 to 300 mg at bedtime. The review concluded that quetiapine may be considered for a limited period (less than 12 weeks) to improve pain, sleep, and depression in fibromyalgia, while considering the potential for weight gain.
A reduction of at least 50% in pain is used by the Cochrane Pain, Palliative and Supportive Care Review Group as a threshold because it correlates with improvements in morbidity, function, and quality of life.
The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the U.S. Army Medical Department or the U.S. Army.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Dean A. Seehusen, MD, MPH, and Randy Bain, DOPublished/Updated
August 1, 2017References:
Tenofovir for Prevention of HIV Infection
Benefits in NNT
50
1 in 50 had HIV infection prevented
Harms in NNT
No one was harmed with serious adverse events
View As:
Source
Okwundu CI, Uthman OA, Okoromah CA. Antiretroviral pre-exposure prophylaxis (PrEP) for preventing HIV in high-risk individuals. Cochrane Database Syst Rev. 2012;(7):CD007189.Study Population: Human immunodeficiency virus (HIV)-negative commercial sex workers, men who have sex with men, and individuals with an HIV-positive partner, mostly in developing settings
Efficacy Endpoints
Incidence of HIV infection; adherence to preexposure prophylaxis (PrEP)Harm Endpoints
Adverse events including renal insufficiency, elevated liver enzymes, nausea, dizziness, and vomitingNarrative
HIV infection occurs in 2.7 million persons each year globally. There remains a need for effective prevention strategies. In 2012, the U.S. Food and Drug Administration approved tenofovir/emtricitabine (Truvada) in the United States for PrEP in conjunction with safer sex practices to reduce the risk of HIV transmission. Nonetheless, research into PrEP has stirred controversy because of concerns about perpetuating risky sexual behaviors, long-term medication effects, and creating drug-resistant strains of HIV from poor compliance.This Cochrane review reports on the results of six trials examining prevention of HIV infection in persons at high risk.1 This review concluded that compared with placebo, tenofovir/emtricitabine (n = 8,918; relative risk [RR] = 0.5; 95% confidence interval [CI], 0.3 to 0.9) and tenofovir alone (n = 4,027; RR = 0.33; 95% CI, 0.2 to 0.6) both significantly reduced the risk of HIV infection. Moreover, serious adverse events were similar for patients in the PrEP and placebo groups, although minor adverse events such as nausea and vomiting were significantly more common in those receiving medications. The Cochrane review does not address the rate of minor adverse events. However, the original studies report rates between 1% and 10% for minor adverse events such as headache, nausea, vomiting, or dizziness.2,3,4
The number needed to treat (NNT) is relatively high because even in these very high-risk groups the annual infection rate was just 4%, perhaps because of safe sex and other prevention education in all groups. The infection rate was roughly 2% in the medication groups, for a raw difference of 2% and an NNT of 50.
Caveats
The studies included here were performed largely in developing countries, where the global new infection burden is highest. It is not yet clear how well this will translate in other settings. Median follow-up varied, typically one to two years; therefore, long-term impact and effectiveness are also unknown. These issues may be better clarified by results from ongoing studies.Tenofovir/emtricitabine is associated with kidney toxicity (reversed when the drug was discontinued in these studies), loss of bone density, and liver inflammation in patients with hepatitis B. There is concern that using tenofovir before transmission of HIV may limit its effectiveness in treating infection later. If persons believe the medication obviates the need for safe behaviors, the intervention could paradoxically increase HIV transmission. A final consideration is cost. As of November 2016, the cost of tenofovir/emtricitabine is approximately $1,000 for a 30-day supply (30 tablets) in the United States.
We have labeled the intervention green (benefits greater than harm), but cost-effectiveness and personal preferences are critical. Treating this target population with HIV drugs so that 2% can avoid infection (and thus avoid the same drugs) may not be a worthy trade-off for some, particularly if adverse events are vexing. For others, the peace of mind offered by taking the medication may be more than worthwhile. The duration of prophylaxis is indefinite or as long as the high-risk exposure continues.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Ben Kaufman, MD, and Shahriar Zehtabchi, MDPublished/Updated
July 1, 2017References:
Efficacy of Direct-Acting Antivirals Compared with Older Agents for Hepatitis C
Benefits in NNT
4
1 in 4 were helped by achieving sustained viral response
Harms in NNT
None experienced major harms
View As:
Efficacy Endpoints
Sustained virologic response (SVR) at 24 weeks (absence of detectable virus in the blood)Harm Endpoints
Anemia, fatigue, death, hospital admissionNarrative
Hepatitis C virus is typically latent (i.e., silent) for decades; therefore, 80% to 85% of persons infected will not experience illness caused by the infection. Conversely, 15% to 20% of patients with untreated hepatitis will eventually develop liver failure or cancer.1 Standard treatment to prevent these developments includes interferon and ribavirin (Rebetol), a difficult-to-tolerate regimen that achieves SVR (i.e., the absence of virus in the blood) in approximately 50% of patients but is commonly associated with anemia and fatigue. The use of newer direct-acting antiviral agents may improve SVR rate and tolerability.2 The review summarized here addresses all major trials of second-generation direct-acting antiviral agents. To better understand the potentially unique benefits of this class, we focus on trials comparing any direct-acting antiviral-based regimen with interferon plus ribavirin. Despite 16 trials being identified, only five (all using simeprevir [Olysio]) were similar enough to be pooled. Simeprevir, when added to interferon and ribavirin, improved SVR without increasing adverse effects: 82% vs. 56% with the older regimen, a 26% absolute improvement (number needed to treat = 4).Although pooled data for other agents such as daclatasvir (Daklinza, used in combination with sofosbuvir [Sovaldi]), ledipasvir (used in combination with sofosbuvir [Harvoni]), sofosbuvir, and paritaprevir (used in combination with ombitasvir and ritonavir [Norvir]) were unavailable (because of their indications to be used in combination with other agents), indirect comparisons suggest similar rates of SVR, typically within 5% to 10%.
Caveats
These data are promising for direct-acting antivirals, and suggest that similar patients have an 80% to 90% chance of SVR when receiving them. This cannot tell us whether these agents prevent liver cancer or liver failure, the goals patients care about, because it is unknown if SVR, a surrogate marker, is adequate to prevent these outcomes or, if so, how consistently. Even assuming SVR leads to universal prevention, the numbers are limited: Up to 20% of infected patients can potentially benefit, and 26% more can achieve SVR with direct-acting antiviral use; therefore, at best, approximately one in 20 patients (0.20 × 0.26 = .05) who receive these antivirals would benefit. Patients with hepatitis C typically present for care because of symptoms or illness. With increased screening, treatment is now commonly considered in otherwise healthy persons who may be at even lower risk of liver problems. Assuming a modest reduction to 10% of untreated patients who would progress to liver failure or cancer, even an overall 82% SVR rate would mean roughly 8% of patients benefit. Comparing this with the 5% to 8% rate of serious adverse events occurring with any treatment regimen suggests that direct-acting antiviral agents—or any other hepatitis C treatment—deserve close examination and shared decision making.3 A recent U.S. Food and Drug Administration report warns about the risk of reactivating hepatitis B virus in patients with current or previous infection who are treated with certain direct-acting antivirals for hepatitis C virus. In a few cases, hepatitis B virus reactivation in patients treated with direct-acting antivirals resulted in serious liver problems or death. Hepatitis B virus reactivation usually occurred within four to eight weeks.4 The studies in this review are relevant only to patients with hepatitis C who have not been treated and do not have cirrhosis. Furthermore, we focused on data from a single agent, simeprevir. Pooled estimates of other direct-acting antivirals were based on “network” meta-analysis, a technique that combines results from a variety of comparisons and settings. This indirect method may be helpful in the absence of head-to-head trials, but we believe the results are less reliable. Network meta-analysis and data from other investigators suggest slightly better results with sofosbuvir.5 Direct-acting antivirals may have introduced a new era in drug pricing. Sofosbuvir, for example, costs $168,000 for a 24-week course, presenting potential barriers to access that have spurred new debates about value and resource allocation. Medicaid reimbursement for sofosbuvir varies, and some states require pretreatment drug screening, abstinence from drugs and alcohol, or suppressed human immunodeficiency virus status, making access a complicated and ongoing issue.6 Direct-acting antivirals for hepatitis C treatment are classified as yellow because more data will be required to determine whether they offer true patient benefit. In particular, follow-up reports on liver cancer and liver failure, as well as trials of hepatitis C screening, are needed. In the meantime, decisions about direct-acting antivirals require thoughtful consideration of benefits and harms, cost-utility, and the limits of financial resources.The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Karen Ta, MD and Shahriar Zehtabchi, MDPublished/Updated
References:
Oral NSAIDs for Perineal Pain in the Early Postpartum Period
Benefits in NNT
3
1 in 3 had adequate pain relief at 4 and 6 hours
5
1 in 5 required no further analgesia after 4 hours
3
1 in 3 required no further analgesia after 6 hours
Harms in NNT
No significant adverse effects
View As:
Source
Wuytack F, Smith V, Cleary BJ. Oral non-steroidal anti-inflammatory drugs (single dose) for perineal pain in the early postpartum period. Cochrane Database Syst Rev. 2016;(7):CD011352.Study Population: Postpartum women who had perineal pain after vaginal delivery and were not breastfeeding
Efficacy Endpoints
Adequate pain relief defined by at least 50% reduction in pain at four and six hours; no additional need for analgesicsHarm Endpoints
Adverse effects at four and six hours, including nausea, vomiting, sedation, constipation, diarrhea, drowsiness, sleepiness, and psychological impactsNarrative
Perineal pain is common after vaginal childbirth. Physicians delivering newborns should be comfortable treating this pain. Oral nonsteroidal anti-inflammatory drugs (NSAIDs) are commonly used in treating postpartum pain and have been studied extensively.1 These medications are not completely benign because they have been associated with adverse cardiovascular, gastrointestinal, and renal side effects.2 With these potential adverse effects in mind, this review evaluated oral NSAIDs for the treatment of perineal pain in the early postpartum period in women who have trauma to the perineum from first- to fourth-degree lacerations.This review included 28 studies that examined 13 NSAIDs and included 4,181 postpartum women. A single-dose NSAID was given to 2,642 women in the early post-partum period, whereas 1,539 received placebo. More women who received an NSAID had adequate pain relief in the first four hours and the first six hours following the intervention compared with those receiving a placebo. Adequate pain relief was defined as at least 50% relief in pain by subjective report or by using the Total Pain Relief score or the Summed Pain Intensity Difference score. Compared with women receiving placebo, those receiving an NSAID achieved adequate pain relief in four hours (relative risk [RR] = 1.91; 95% confidence interval [CI], 1.64 to 2.23) and in six hours (RR = 1.92; 95% CI, 1.69 to 2.17). The numbers needed to treat to prevent use of additional analgesics at four and six hours were 5 and 3, respectively. Women receiving an NSAID had less need for additional analgesia at four hours compared with placebo (RR = 0.39; 95% CI, 0.26 to 0.58) and six hours (RR = 0.32; 95% CI, 0.26 to 0.40). Only one study recorded adverse events in the four-hour post-intervention group with very small numbers. Compared with placebo, those receiving an NSAID had no difference in overall adverse events (RR = 1.38; 95% CI, 0.71 to 2.70). These adverse events included nausea, vomiting, sedation, constipation, diarrhea, drowsiness, sleepiness, and psychological impacts.
Caveats
This review included only randomized controlled trials. Most of the studies were completed in the 1980s and had a wide range of sample sizes, patient types, and treatment settings. The individual studies had poor reporting of bias, making it difficult to ascertain the level of bias. As a result, the quality of evidence in the individual studies ranged from very low to low grades.Neonatal outcomes were not evaluated in any of the studies. Finally, including non-breastfeeding women could be considered a limitation as well. The authors of the Cochrane review could not identify a physiologic reason to exclude them from the study, but assumed they were excluded because many of the studies were published in the 1980s before much of the evidence supporting breastfeeding was discovered and published. There is no clear reason to think these results do not apply to breast-feeding mothers as well.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Dean Seehusen MD, MPH, and Tyler Rogers, MDPublished/Updated
May 1, 2017References:
Vitamin E in Pregnancy
Benefits in NNT
333
1 in 333 avoided a placental abruption
Harms in NNT
19
1 in 19 developed term premature rupture of membranes
No change in neonatal death, infant death, or preterm birth rates
View As:
Source
Rumbold A, Ota E, Hori H, Miyazaki C, Crowther CA. Vitamin E supplementation in pregnancy. Cochrane Database Syst Rev. 2015(9):CD004069.Study Population: Pregnant women
Efficacy Endpoints
Placental abruption avoidedHarm Endpoints
Stillbirth, neonatal death, perinatal infant death, preterm birth, preeclampsia, intrauterine growth restriction, premature rupture of membranes (PROM), placental abruptionNarrative
Vitamin E has antioxidant properties that decrease oxidative stress within the body. During pregnancy, increased oxidative stress has been linked to pre-eclampsia, intrauterine growth restriction, and PROM. This review evaluates the effectiveness of vitamin E supplementation in pregnancy.1Approximately 22,000 women from 17 trials were included in the analysis. The review reports no clear difference in stillbirth or neonatal death. There was also no difference noted for perinatal death, preterm birth, preeclampsia, or intrauterine growth restriction, although the authors note substantial heterogeneity for these specific outcomes.
PROM and placental abruption were considered secondary outcomes. Heterogeneity is a limiting factor for analysis of preterm PROM; however, the authors reported no difference using advanced data analysis. There is a statistically significant increase in the risk of term PROM in patients taking vitamin E supplements vs. the control groups (number needed to harm = 19; P < .001). Patients who received vitamin E had a decreased risk of placental abruption (number needed to treat = 333; P = .02).
Among the most patient-important outcomes, such as infant death, vitamin E does not show any benefit. Although there may be some benefit in decreasing placental abruption, vitamin E may cause harm during pregnancy by increasing term PROM.
Caveats
Perhaps the most notable limitation, beyond the marked heterogeneity, is the administration of vitamin E with other supplements. All of the studies included in the review gave patients vitamin E with other supplements, usually vitamin C. This complicates the data because of the inability to separate the effects of vitamin E from the effects of the other supplements.Vitamin E supplementation is a controversial topic in newborns, increasing the importance of these data. Oxidative stress in newborns is implicated in intraventricular hemorrhage, retinopathy, and lung disorders. Vitamin E supplementation in newborns for prevention of these complications has been associated with an increase in neonatal sepsis and necrotizing enterocolitis.2 This review does not support the use of maternal vitamin E supplementation because of complications with the pregnancy but does not look at the rate of newborn outcomes such as intraventricular hemorrhage. Death rates in the newborn were analyzed, but no statistically significant difference was noted.
Based on available data, further studies focused on vitamin E supplementation alone are not warranted and may not be safe for the pregnant patient.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Patricia Van Leer, MDPublished/Updated
April 1, 2017References:
Buprenorphine Maintenance vs. Placebo for Opioid Dependence
Benefits in NNT
4
1 in 4 using low-dose buprenorphine (2 to 6 mg) had retention in treatment
3
1 in 3 using medium-dose buprenorphine (7 to 16 mg) had retention in treatment
2
1 in 2 using high-dose buprenorphine (≥ 16 mg) had retention in treatment
Harms in NNT
No study-related medication mortality was reported
Uncertain adverse effects
View As:
Source
Mattick RP, Breen C, Kimber J, Davoli M. Buprenorphine maintenance versus placebo or methadone maintenance for opioid dependence. Cochrane Database Syst Rev. 2014;(2):CD002207.Study Population: Adults with opioid dependence
Efficacy Endpoints
Treatment retention and illicit drug use suppressionHarm Endpoints
Mortality and adverse effectsNarrative
The United States is facing an opioid epidemic. Since 1999, overdose deaths involving opioids and heroin have quadrupled.1 Methadone, a full agonist, has traditionally been used to treat opioid and heroin dependence. Buprenorphine, a partial agonist, is an appealing alternative because it causes lower physical dependence, milder withdrawal symptoms, and is less likely to cause an overdose.2 This review examines the effectiveness of buprenorphine vs. placebo for retention in treatment and illicit opioid use, as measured by positive urinalysis or self-reported use.This Cochrane review included 31 studies with 5,430 participants, the majority of whom were male with an average age of 30 years.2 High-quality evidence shows that buprenorphine at all doses is more effective than placebo in retaining patients in treatment. An analysis of five studies using low-dose buprenorphine (2 to 6 mg) in 1,131 participants showed a benefit in treatment retention over placebo (relative risk [RR] = 1.5; 95% confidence interval [CI], 1.19 to 1.88). Medium-dose buprenorphine (7 to 16 mg) had slightly better patient retention in treatment (RR = 1.74; 95% CI, 1.06 to 2.87). High-dose buprenorphine (≥ 16 mg) was the most effective at retaining study participants in treatment (RR = 1.82; 95% CI, 1.15 to 2.90).
This review found moderate-quality evidence that buprenorphine is effective in suppressing illicit drug use, but only at doses of 16 mg or greater (three studies; 729 participants; standardized mean deviation [SMD] = −1.17; 95% CI, −1.85 to −0.49; number needed to treat = 2). Low-dose and medium-dose buprenorphine did not suppress illicit opioid use (SMD = 0.10; 95% CI, −0.80 to 1.01 and SMD = −0.08; 95% CI, −0.78 to 0.62, respectively).
Only five studies reported mortality data; no deaths were reported in three of the studies. In two of the studies there was a 20% mortality rate reported in the control group. Two deaths unconnected to the study medication were reported in one trial due to stab wounds and cancer.
Caveats
All trials included were randomized, controlled studies with low risk of bias. Twenty-two were double-blinded and 10 were open comparative trials. Of note, four of the studies used a 1-mg dose of buprenorphine as the placebo. The authors note that this is unlikely to bias results given the clinically insignificant dose. The included studies varied in their reporting of adverse events, if they reported them at all.Current treatment of opioid dependence generally uses an individualized dosing schedule; therefore, flexible-dose methadone or flexible-dose buprenorphine may be more clinically relevant. This review also compared 11 studies with 1,391 participants receiving either flexible-dose methadone or flexible-dose buprenorphine. The methadone group was more likely to be retained in treatment (RR = 0.83; 95% CI, 0.73 to 0.95). There was no difference between the two groups with regard to illicit opioid use (SMD = −0.11; 95% CI, −0.23 to 0.02).
Conclusion: Based on current evidence, buprenorphine is effective in retaining patients in treatment at all doses; however, only doses of at least 16 mg prevent illicit drug use during therapy. Studies using flexible-dose buprenorphine without using buprenorphine as a placebo may be more clinically relevant to actual practice. The harms from treatment are currently unclear because of poor reporting of adverse events.
The views expressed are those of the author and do not reflect the official policy or position of the Bayne-Jones Army Community Hospital, the Army Medical Department, the Department of Defense, or the U.S. government.
The original manuscript was published in Medicine by the Numbers, American Family Physician as part of the partnership between TheNNT.com and AFP.
Author
Meghan F. Raleigh, MDPublished/Updated
March 1, 2017References:
Glatiramer Acetate (Copaxone) for Multiple Sclerosis
Benefits in NNT
8
1 in 8 with relapsing-remitting MS were helped (had a relapse prevented within the first year)
None saw an improvement in progression of disease